
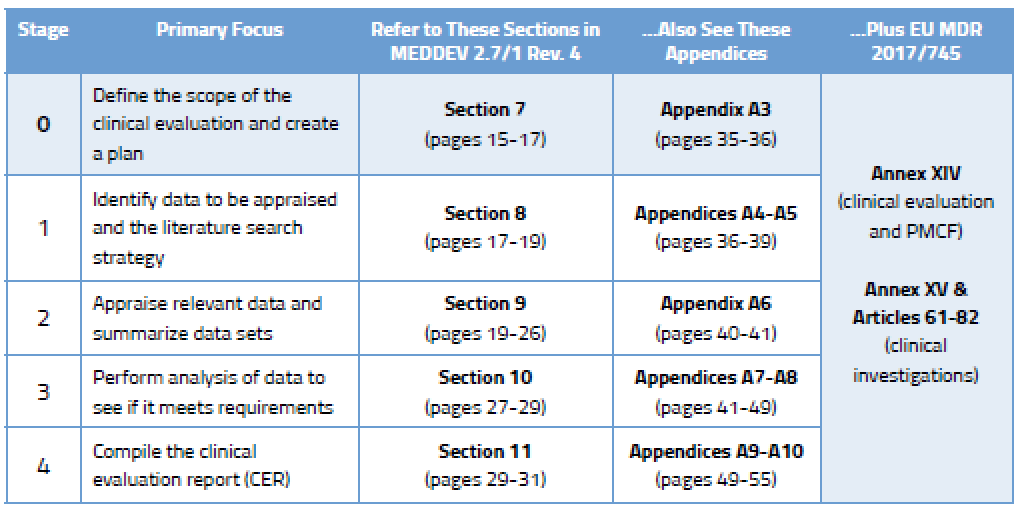
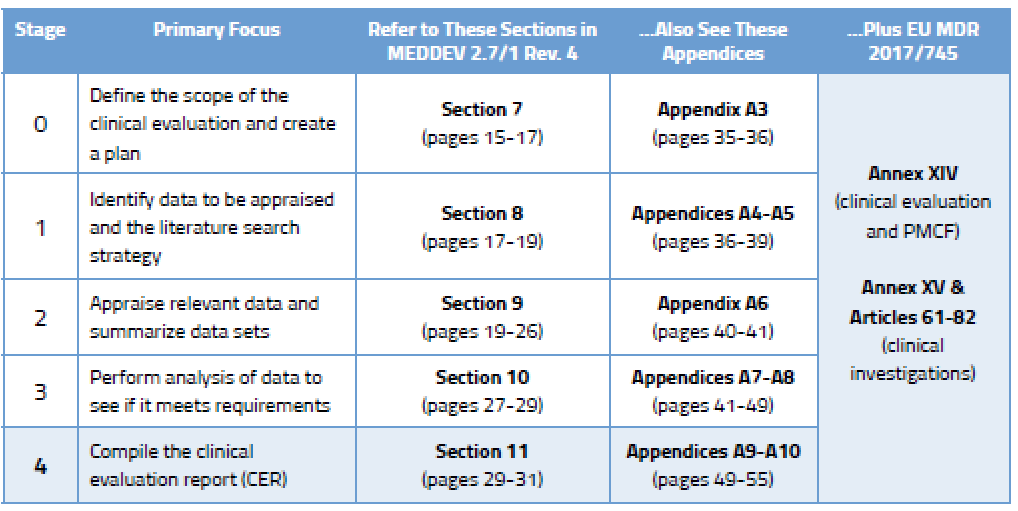
Before we go into detail on the process, here’s an overview of the various stages outlined in MEDDEV 2.7/1 Rev. 4 and where you will find guidance on each stage.
There are two broad categories of clinical data.
1 – Data not generated by your company (retrieved from online literature searches and other offline data)
2 – Data generated by your company, which can include:
During Stage 0, you came up with a clinical evaluation plan focused on determining which data you need and from what sources. In Stage 2, your focus will turn to choosing the right data among the data sets you have found. This requires yet more planning. Your data appraisal plan should address:
According to Section 9.2 of MEDDEV 2.7/1 Rev. 4, the criteria you select should “reflect the nature, history and intended clinical use of the device.” This is something you must document and justify based on current state of the art.
During Stage 3 you are expected to analyze the data to ensure the clinical evaluation demonstrates that any risks are minimal and acceptable. You also need to consider all aspects of the device’s intended purpose. You’ll find more detail on this in Section 10.2 of MEDDEV 2.7/1 Rev. 4, and in Annex AVII. You will identify gaps related to:
Data from the literature you have appraised is often put into Excel tables to be analyzed. It’s a convenient way to compare different study details, patient populations, endpoints, adverse events, etc. This can end up being a sizable amount of data unto itself, but certain aspects can be parsed into “bite-sized” tables focused on particular issues. While spreadsheets are not especially wonderful for narrative explanations, they are useful as quick comparative tools and are extremely helpful in noting differences between studies when writing the summary.
Section 6.2.3 of MEDDEV 2.7/1 Rev. 4 provides guidance to manufacturers on how often to update clinical evaluations. It says that the “manufacturer should define and justify the frequency” of CER updates. Typically, this is done in concert with your Notified Body audit and certificate renewal, but that predefined schedule can be tossed out the window if your postmarket surveillance activities uncover new risks.
Shown below is an example of how you could rationalize the frequency of updates to your CER. This sample shows a simple evaluation table for an ECG machine that is well established on the market and has decent clinical history. You can, of course, create your own rating system with more factors, and you could weight these factors as well. We’ve kept the table simple in order to illustrate the point that your NB wants to see that you have a systematic approach to determining your CER update schedule.
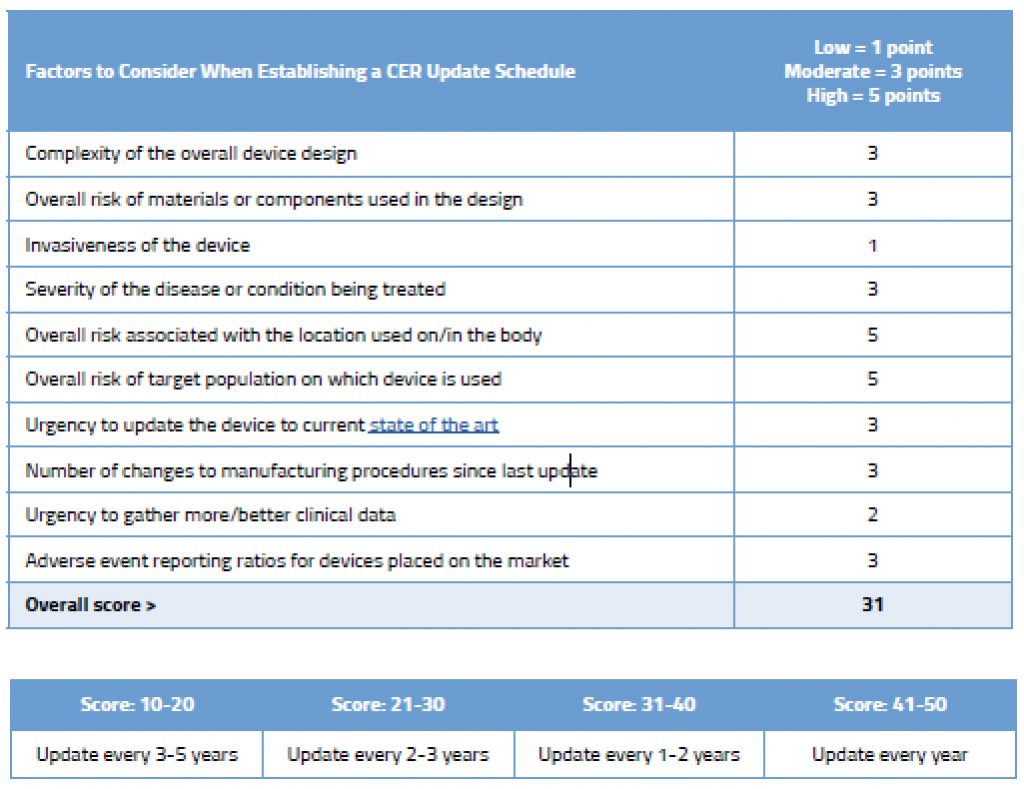
As you can see in this example, even though this device is an established ECG with good clinical history, due to its inherently complexity, where it is used on the body, and the target population on which it is used, updating the CER every few years is still recommended.
Regardless of your proposed rationalization for the update schedule, you must update the CER after getting new information from your postmarket surveillance activities that might change your current evaluation. For example, safety reports, newly published literature, or PMCF studies may uncover previously unknown safety concerns. Data from these sources must be to evaluated because it may change your risk/benefit profile. Remember that Annex XIV, Part B of the MDR mandates that your PMCF plan specify the “methods and procedures for proactively collecting and evaluating clinical data.”

US OfficeWashington DC
EU OfficeCork, Ireland


