Jul 15, 2026
Why FDA Inspections Feel Different Under QMSR: Part II

Jul 01, 2026
Why FDA Inspections Feel Different Under QMSR: Part I

Jun 26, 2026
QMSR Is Taking the Spotlight, But MDSAP Still Deserves Attention

Jun 26, 2026
Beyond the Checklist: What Effective Good Laboratory Practices (GLP) Auditing Really Looks Like

Jun 24, 2026
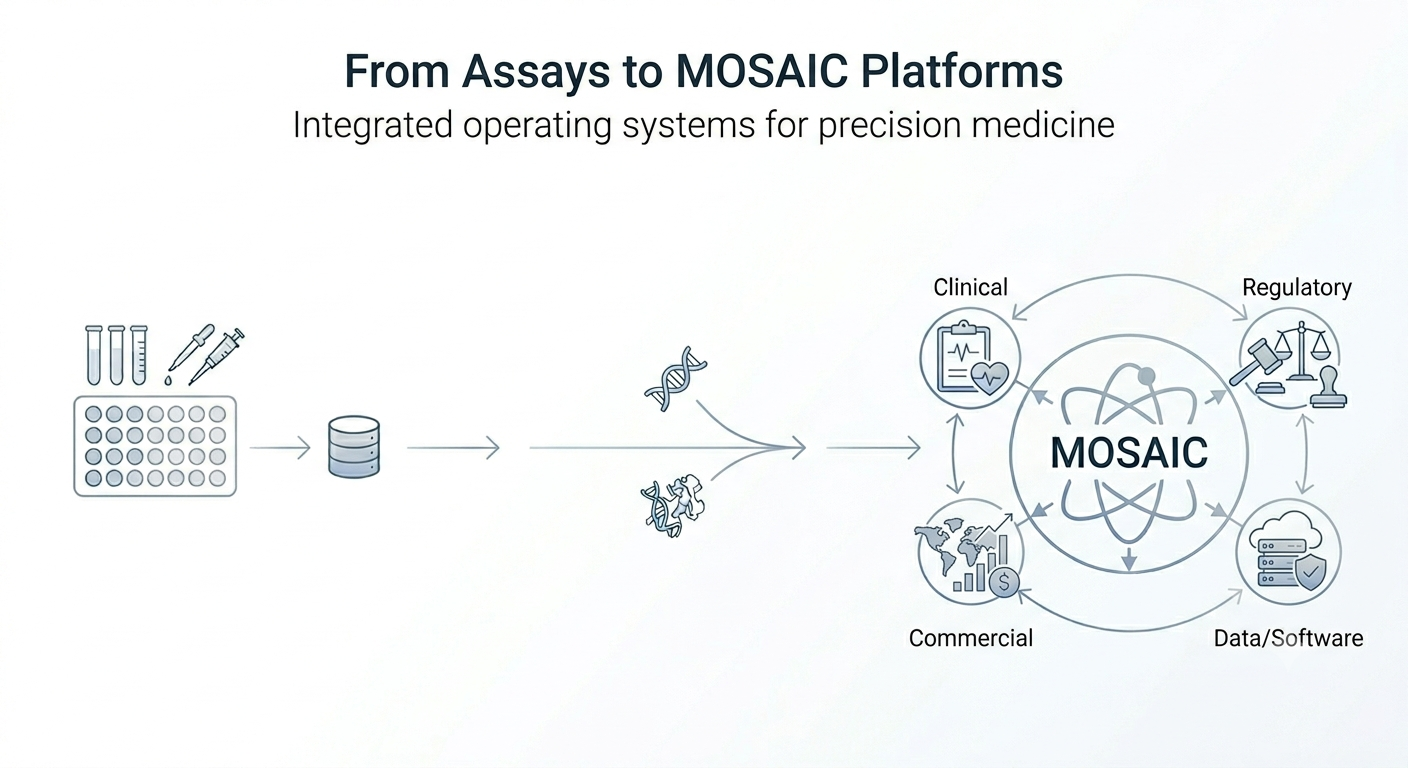
From Assays to MOSAIC Platforms: The Next Operating Model for Precision Medicine
Jun 05, 2026
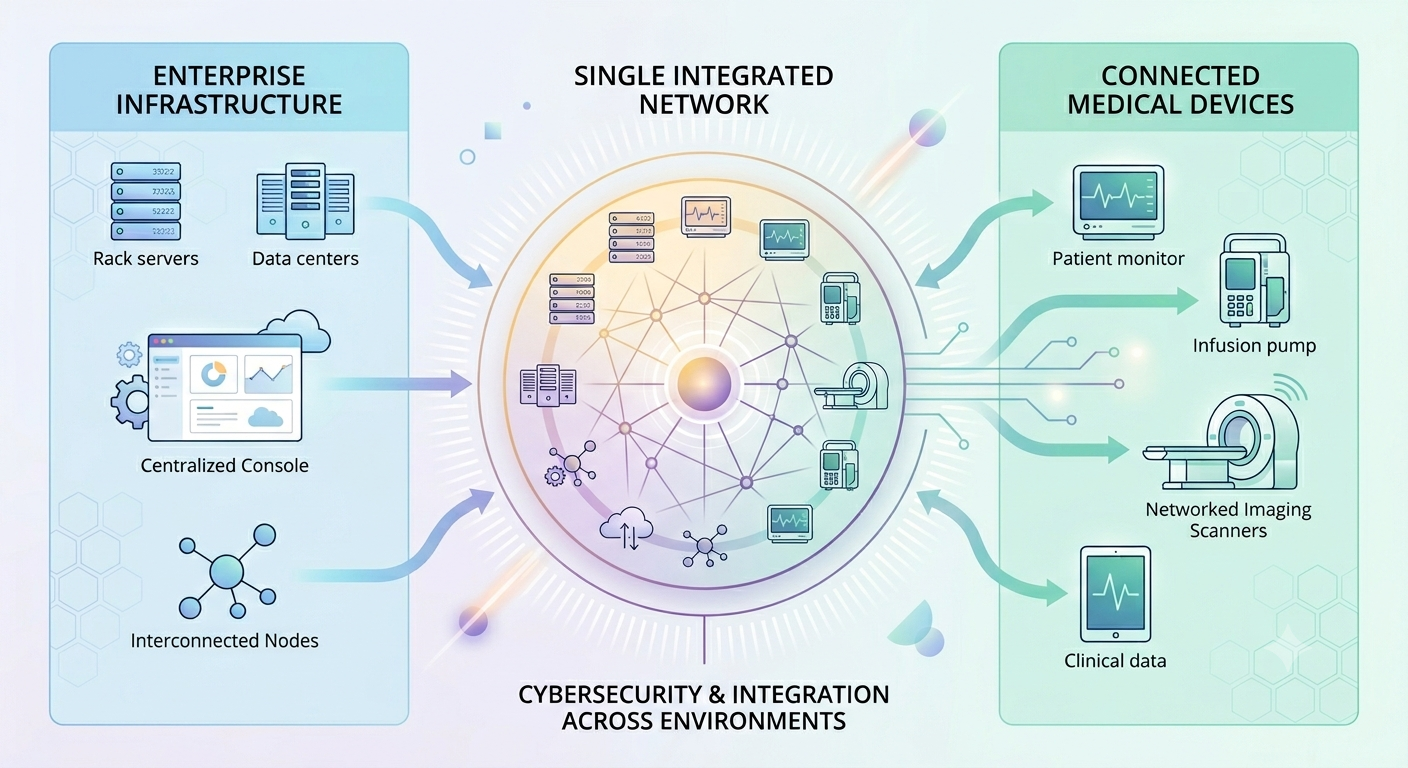
Medical Device Cybersecurity: Bridging Enterprise Security and Product Security