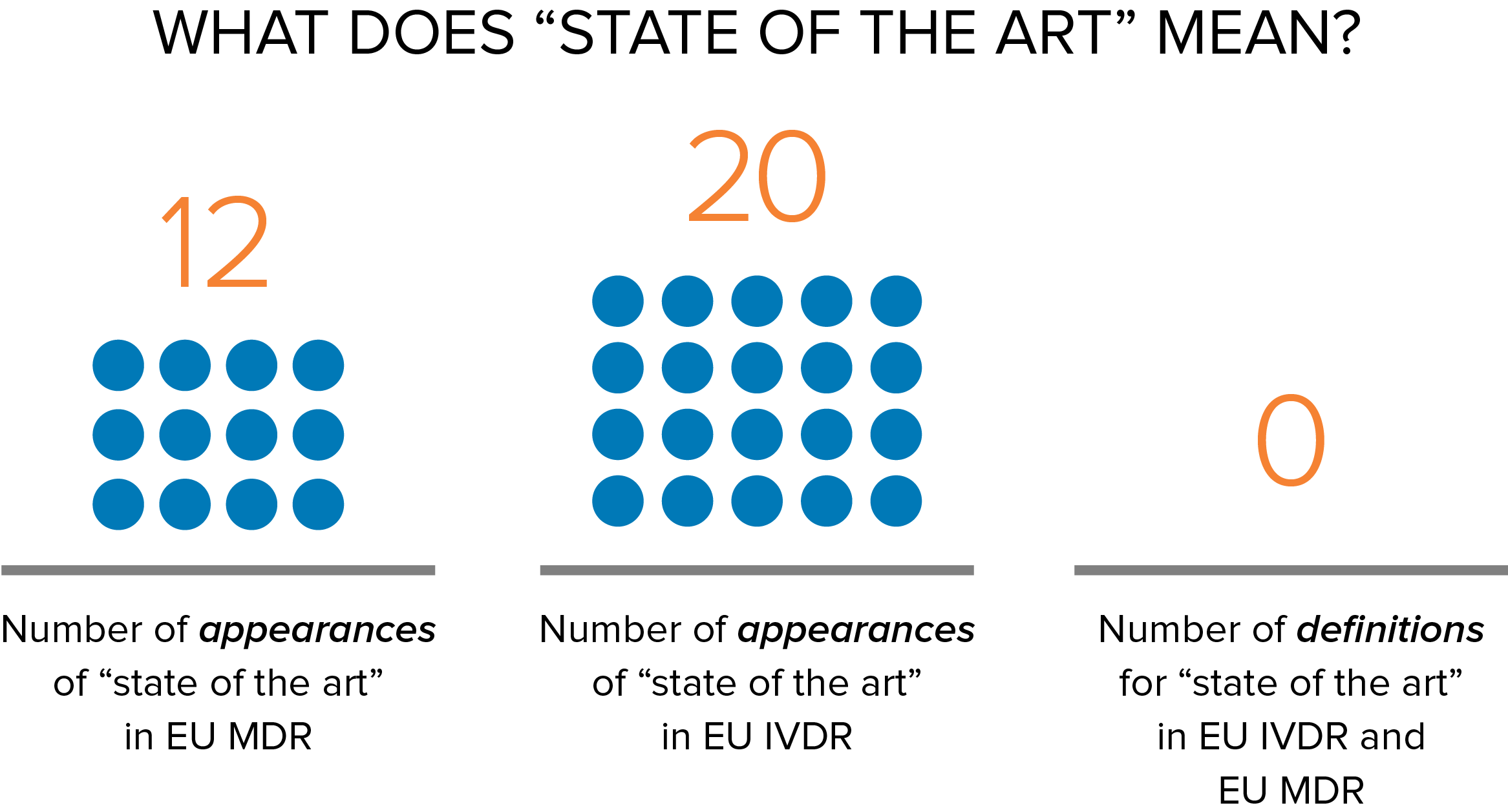
Confused? It’s understandable. Most of us think of state of the art as describing the latest and most advanced stage of a technology. For example, most people would consider a bionic eye to be state of the art. Yet, this notion of state of the art isn’t what is meant in the context of the EU MDR, IVDR, and risk management. As such, manufacturers often face challenges in assessing state of the art in design and postmarket surveillance. Some of the confusion arises because, in the medical device and IVD regulatory arena, state of the art refers only to products that are developed and approved for sale in the marketplace, not cutting-edge technology in development. As we all know, there is a very big difference between a new state-of-the-art digital imaging system that is undergoing trials and one that already has CE Marking. For EU medical device regulators, the latter is considered state of the art, but the former is not until it has CE Marking.
Here’s one way to think about this. Ask yourself: What is state of the art in cars these days? You might think of self-driving cars. But the reality is that while self-driving cars are highly innovative, they don’t represent the widespread commercially available stage of technology. In the context of the EU Medical Device Regulation, state of the art is really the middle of a bell curve in innovation. A Model T certainly isn’t state of the art, but cars with side curtain airbags, Bluetooth and backup cameras could be considered state of the art under the ISO 14971:2019 3.28.
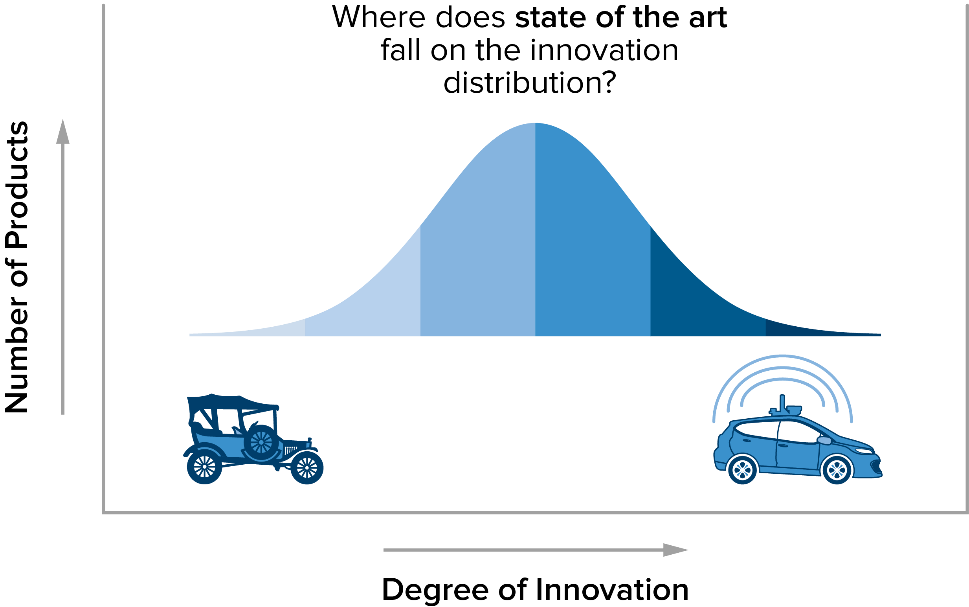
A note associated with this definition from ISO 14971:2019 3.28 adds more insight: “The state of the art embodies what is currently and generally accepted as good practice. The state of the art does not necessarily imply the most technologically advanced solution.” Thus, it is more useful to think of state of the art as meaning “the current state of all competitive treatment options.”
If your medical device or IVD is not the most technologically sophisticated product on the market but has a sparkling safety record, possesses solid clinical data, and follows all the latest standards, does that mean it still meets best practices and qualifies as state of the art in the eyes of EU regulators?
Maybe.
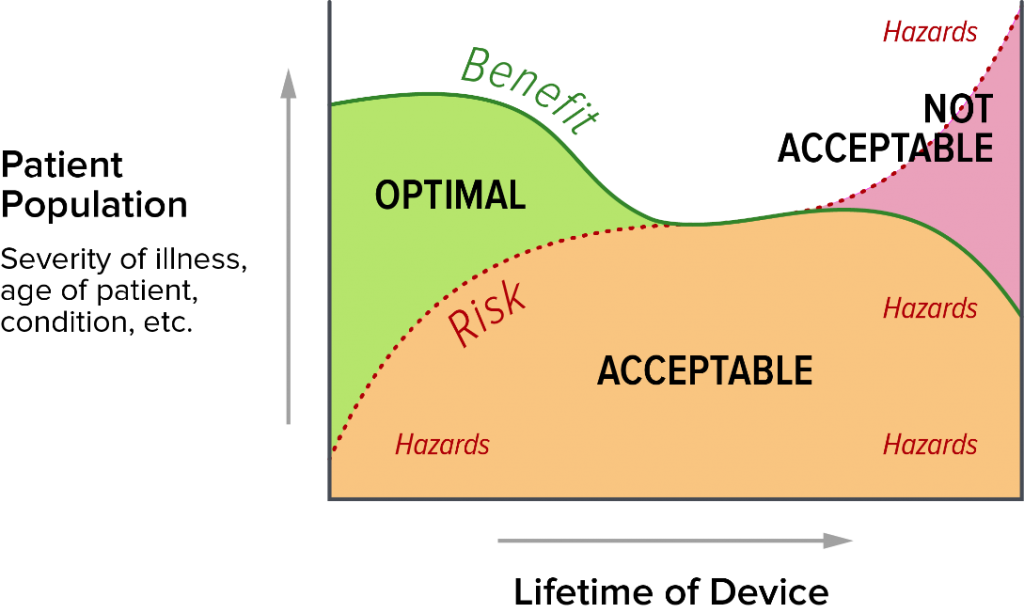
It also depends on the benefit-risk balance over the life of the device. As the device ages, assuming no new versions are introduced, it becomes less state of the art (like a Model T). It also has less benefit as newer treatment options are developed. Even if the risk profile is unchanged for your device, there will be less risky devices on the market.

US OfficeWashington DC
EU OfficeCork, Ireland


