QA/RA Consulting, Auditing & Training
Since the introduction of the In Vitro Diagnostic Directive (IVDD 98/79/EC) 20+ years ago, the majority of IVD manufacturers selling in Europe have had it pretty easy when it comes to EU compliance. That’s because under IVDD 98/79/EC, about 80-90% of finished devices are not subject to Notified Body oversight. However, the era of laissez faire is coming to an abrupt end.
European regulators have long recognized the shortcomings of the existing IVDD in protecting patients and spent years crafting an updated and greatly expanded set of regulations known as the In Vitro Diagnostic Regulation (2017/746), or simply the IVDR. To underscore the importance of stricter oversight, the requirements governing IVDs have been elevated to a regulation from a directive, which means every EU member country must apply the same regulation in their national law, with the same implementation date.
The IVDR is not simply a fresh coat of paint for the IVDD. EU regulators have torn down the existing building and reconstructed it using new blueprints. The IVDR greatly increases the number of IVD devices subject to rigorous oversight and expands the scope of compliance. It is based on the GHTF classification system and brings European regulations into closer harmonization with global classification for in vitro diagnostic devices that has been around for many years. To get a sense of the magnitude of the changes, simply look at the table below. The IVDR is roughly 4x longer than the IVDD it will replace!

Throughout this article series we will reference numerous IVDR Annexes and Chapters. Navigating the IVDR can be quite a challenge. The crafters of the IVDR – having already succeeded at giving heart palpitations to thousands of IVD RA/QA professionals – inflicted a final wound by leaving out a Table of Contents to navigate their 157-page tome. Fortunately, we have saved you from endless frustration and created a Table of Contents for you! You can download a fully linked IVDR Table of Contents, which includes the original IVDR text.
It is obviously impossible to cover every change and requirement found in the IVDR, but here’s a high-level summary of the most important updates. We’ll talk about these in more detail later.
Not long ago, the definition of an IVD in Europe was pretty clear-cut. Today, not so much. As new diagnostic technologies have developed, the traditional concept of in vitro diagnostics has expanded. Health data that was previously delivered only through clinicians is now available directly to patients on their smartphones or via email. Advances in genetic testing have made predictive health data available to the masses. That same technology can be used to help customize treatment plans for patients. Such advances offer obvious benefits for patient care, but they also bring the risks associated with false-positives and misinterpretation of data. Given these ongoing advances, the IVDR expands the previous definition of an in vitro diagnostic medical device, as seen in the highlighted text below.

The current IVD directive uses a list-based classification scheme that is very limited in application. In fact, that list in Annex II of the IVDD only takes up half a page, with a short list of what is reviewed by Notified Bodies. The IVDR throws those lists out the regulatory window and applies a new risk-based classification system that’s based heavily on a GHTF IVD classification guidance published in 2008.
Article 47 of the IVDR briefly discusses classification, but Annex VIII contains the seven rules you will use to classify your device. The new IVDR separates devices into four classes: A, B, C, and D. Class A is for low-risk products and Class D, as you smartly surmise, is reserved for high-risk products.
Under the new scheme, the vast majority of in vitro diagnostics that will require Notified Body intervention will fall under Classes B and C. Class D IVDs are quite limited in number, as is the case with Class III devices under the MDR. However, it should be noted that sterile IVDs in Class A will also require Notified Body assessment, although only pertaining to the sterilization aspects. The classification rules also bring companion diagnostics (CDx), genetic testing, and stand-alone software into the gravitational orbit of the IVDR. If the device has multiple intended uses – and those uses place the device into different classes – the highest applicable classification applies.
Structure of the In Vitro Diagnostic Regulation (2017/746) Download the complete IVDR plus a linked Table of Contents. | |
Chapter I | Introductory provisions |
Chapter II | Making available on the market, and putting into service of devices, obligations of economic operators, CE Marking, free movement |
Chapter III | Identification and traceability of devices, registration of devices and economic operators, summary of safety and clinical performance, European database on medical devices |
Chapter IV | Notified bodies |
Chapter V | Classification and conformity assessment |
Chapter VI | Clinical evidence, performance evaluation and performance studies |
Chapter VII | Postmarket surveillance, vigilance and market surveillance |
Chapter VIII | Cooperation between member states, medical device coordination group, EU reference laboratories and device registers |
Chapter IX | Confidentiality, data protection, funding and penalties |
Chapter X | Final provisions |
Annex I | General safety and performance requirements |
Annex II | Technical documentation |
Annex III | Technical documentation on postmarket surveillance |
Annex IV | EC declaration of conformity |
Annex V | CE Marking of conformity |
Annex VI | Information to be submitted upon the registration of devices and economic operators in accordance with articles 26(3) and 28, core data elements to be included in the UDI database together with UDI-DI in accordance with articles 25 and 26 of the UDI system. |
Annex VII | Requirements to be met by Notified Bodies |
Annex VIII | Classification rules |
Annex IX | Conformity assessment, based on a quality management system and on assessment of technical documentation |
Annex X | Conformity assessment based on type examination |
Annex XI | Conformity assessment based on production quality assurance |
Annex XII | Certificates issued by a notified body |
Annex XIII | Performance evaluation, performance studies and postmarket performance follow-up |
Annex XIV | Interventional clinical studies and certain other performance studies |
Annex XV | Correlation table (with the IVDD) |
The timelines for transition from the In Vitro Diagnostic Directive (98/79/EC) to the In Vitro Diagnostic Regulation (IVDR 2017/746) are outlined in Article 110 of the IVDR. However, the EU Parliament and European Commision adopted an amendment to provide longer IVDR transitions period for most, but not all, IVDs. See below the new IVDR transition timelines by device classification.
Class A non-sterile IVD – Must comply with IVDR immediately
Class A sterile IVD – Must comply with IVDR by May 26, 2027
Class B IVD – Must comply with IVDR by May 26, 2027
Class C IVD – Must comply with IVDR by May 26, 2026
Class D IVD – Must comply with IVDR by May 26, 2025
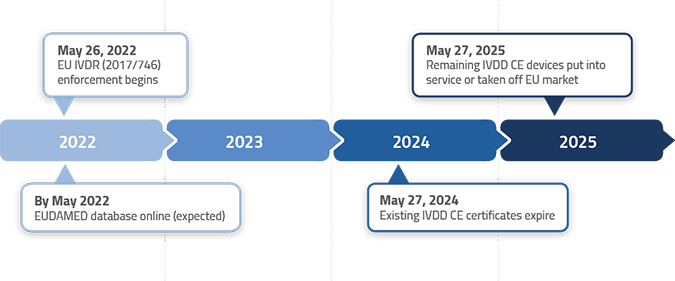
Here are some typical examples of devices and when they might/must make the transition from IVDD to IVDR
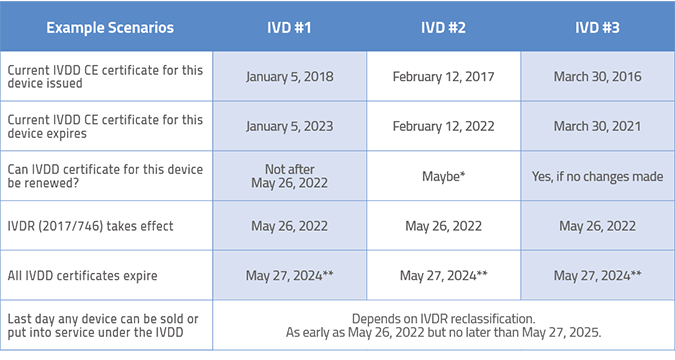
*Because the renewal date for this IVDD certificate falls so close to the IVDR implementation date, it may be difficult to get Notified Body resources for renewal. Plan far ahead.
**Even though the certificate is normally valid for 5 years, the IVDR mandates that all IVDD certificates expire no later than May 27, 2024.
Depending on your IVDR classification, the last day any device can be sold or put into service under the IVDD is as early as May 26, 2022 but no later than May 26, 2027.
Of course, the table above focuses on how you can maximize the leniency associated with the existing In Vitro Diagnostic Directive (98/79/EC). In some cases, you may choose not to renew your existing IVDD certificate and pursue IVDR certification instead. That may seem crazy, but there are situations where this may make sense. If your product has strong clinical data, certify early to gain a competitive advantage. If it has a marginal story to tell, perhaps wait to allow more time to gather the necessary data. Smart companies will be rewarded with lower costs if they take this seriously and do not procrastinate on their compliance efforts.
As mentioned, IVD classification is changing; as a result, many more devices will require review by Notified Bodies. This means European Notified Bodies (NB) will review your compilation of technical information, classification rationale, and – more important – your documentation supporting performance evaluation. A significant number of IVD companies that sell in the United States may find it easier to achieve compliance with the EU IVDR than companies that do not sell those same IVDs in the US. Why? Companies selling diagnostic products that are 510(k) cleared in the US will already have obtained from clinical sources much of the supporting testing and performance data that is necessary to comply with many (but certainly not all) of the new IVDR requirements. Of course, there are many IVD products that are 510(k)-exempt and not subject to FDA scrutiny (e.g., common blood tests), so companies need to carefully examine the new EU IVDR classification rules in Annex VIII to see where the chips fall.
Let’s take a look at the technical documentation requirements. We will talk more about specific postmarket data requirements later.
Most manufacturers of Class B, C, and D devices will generally need to provide the following information for Notified Body review in order to obtain CE Marking. Manufacturers of Class A devices also need to compile this information, but it will not be reviewed by a Notified Body prior to those devices being placed on the EU market.
Technical documentation includes:
Clinical performance can be demonstrated through:
Clinical testing to generate performance data will be required for Class B, C, and D in vitro diagnostic devices (and Class A as applicable) and the requirements are more complex than they are for most medical devices. As the manufacturer, you must demonstrate that your test is necessary to make a medical decision related to indications for use established for the device. For manufacturers of established and standardized tests, putting together this information will be easier if clinical guidelines are already in place, similar tests exist in the marketplace, and comparable results exist regardless of manufacturer.
Save time and frustration! Download a fully linked IVDR Table of Contents that includes the original IVDR text.
The PER is new to the IVDR but is similar to the EU Clinical Evaluation Report (CER), which is common in the medical device world. The PER is not a single report; rather, it is a collection of data that forms a key element of your overall technical documentation. Chapter VI, Article 56 and Annex VIII, Part A (1.3.2) of the IVDR outline the specific components of the PER and specify that it must include the following:
It is important to keep in mind that the Performance Evaluation Report is not a “once and done” exercise! The IVDR makes it very clear that you are expected to update your PER throughout the entire life cycle of the device with data from your Postmarket Performance Follow-Up (PMPF) plan. Performance reports for Class C and D devices must be done as needed but at least annually. PERs for Class A and B devices can be updated as needed, although if you make Class B devices and have not updated your PER in three years, you better have a good explanation as to why it may not be current when your Notified Body comes knocking during your surveillance audit.
The PER has all of the analytics like sensitivity, specificity, ROC curves, reproducibility, stability, etc. These have been required as part of the FDA 510(k) process for a long time, so those companies that have secured US clearance/approval will find the PER reasonably easy to compile. However, like the CER required for medical devices sold in Europe, the PER must support the clinical benefit and clinical utility of the IVD test. This means that a manufacturer may have a test with an intended use to find biomarkers Beta42, tau-131, and tau-233 that claims if all three are present then there is an 85% chance of developing Alzheimer’s. Your PER must have the clinical data to support a claim like this.
In addition to the requirements noted above, Annex IX, Section 5.1 also requires manufacturers of Class B, C, and D self-testing and near-patient testing devices to submit their product (unless impractical) for a design evaluation by the Notified Body. After this physical assessment of your product, your Notified Body may ask you to carry out more tests. Section 5.2 stipulates specific requirements for companion diagnostics (CDx), so be sure to read this section in Annex IX if that applies to you.
The European Commission published MDCG 2022-2 on guidance for compiling a Performance Evaluation Plan and a Performance Evaluation Report. All of this documentation must be made permanently available to your European Authorized Representative (EC REP). Additional European Commission MDCG endorsed documents and guidances in applying IVD regulations can be found at MDCG endorsed documents and other guidance.
Article 15 of the IVDR introduces the new role of the Person Responsible for Regulatory Compliance (PRRC). Make sure you read this IVDR Article (and this article) carefully. This appointee (perhaps you!) will have tremendous responsibility and must meet certain educational and/or professional experience requirements. Note that the PRRC has different responsibilities than your Authorized Representative (EC REP).
Annex X, Section 3c sets the tone for how seriously regulators will be looking at your clinical data as part of performance evaluation. It states: “The notified body shall employ device reviewers with sufficient clinical expertise and, if necessary, use external clinical experts with direct and current experience relating to the clinical application of the device in question for the purposes of that review.”
That does not mean that Notified Bodies will be reviewing technical documentation for every device in your arsenal. For Class B devices, one set of technical documentation will be evaluated per device category. For Class C devices, one per generic device group. You should expect a different device to be evaluated each year. Notified Bodies will use random sampling as a proxy for your overall state of compliance. Because the Notified Body gets to choose, you better make sure all of your devices are compliant. Class D, companion diagnostics (CDx), self-testing devices, and point-of-care devices are the exceptions to the sampling rule – all technical documentation for such devices will be evaluated.
Your technical documentation must be kept on file for at least 10 years from the date the last product was placed on the European market. Chapter III of Annex IX outlines the specifics.
The IVDR will affect companies in vastly different ways. Some will be in good shape and have most or all of the necessary clinical data. Some, however, will be unpleasantly surprised. If your company makes an IVD that:
… then buckle up – you are in for a wild ride over the next few years. The regulatory landscape is set to change dramatically in Europe and the carefree days of minimal EU regulatory oversight (for most) are nearly over. Sometime in the (near) future you may find yourself around a conference room table with other managers discussing whether the new IVDR paradigm is worth the effort for specific products in your portfolio. Go into that meeting with eyes wide open.
The European IVDR mandates that all manufacturers – even those who manufacture Class A products – have an operating QMS. While manufacturers of all Class A products need a modest QMS and may “self-declare” conformity with CE Marking, there is no free pass for manufacturers of Class B, C, and D products. Most companies currently in compliance with the FDA Quality System Regulation (21 CFR Part 820) will need to upgrade their systems to comply with ISO 13485:2016 if they have not already done so. Even though ISO 13485 is technically a voluntary standard for QMS compliance, in reality it is the de facto standard for QMS compliance.
Manufacturers of Class C and D devices have options for conformity assessment in the IVDR as follows:
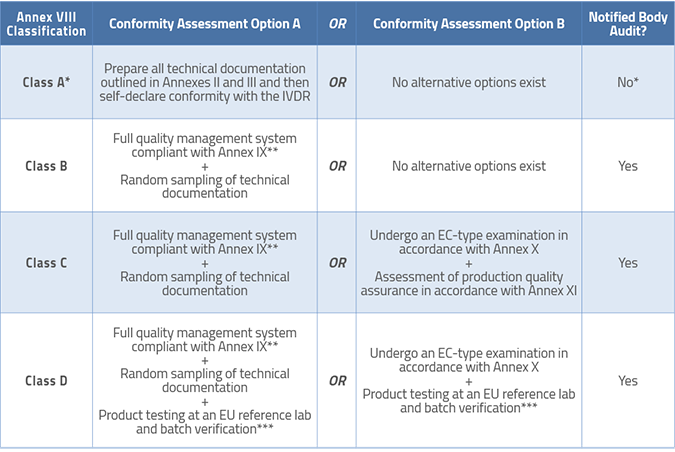
Article 46 mandates that all Notified Bodies must post their standard fees for performing conformity assessments, so it should be easy for you to compare prices as you prepare your compliance plan and budgets.
Chapter VII of the IVDR outlines which type of data, sources, and analysis methods manufacturers should collect as part of ongoing efforts related to postmarket activities. The purpose is to gather postmarket data as part of a manufacturer’s continuing review of risk versus clinical benefit associated with their products. The chapter is actually quite similar to what is required for medical devices under the MDR (EU 2017/745). This includes the need for manufacturers to:
Annex III provides some additional detail on the technical documentation needed related to postmarket surveillance. Notable among the requirements is that you must maintain a “proactive and systematic process” of collecting information. Annex III also mandates that you prepare a Postmarket Performance Follow-Up Plan (PMPF) or provide a reason why it should not apply to your device.
An important thing to understand about postmarket surveillance under the IVDR is that your PMS process must be continuous and proactive (not static and reactive) and has a direct link to your risk management file. Gone are the days when you could market your IVD in Europe and assume all was well because no incidents were reported last year. Your Notified Body will be examining your efforts to proactively monitor the safety of your products on the EU market. Postmarket surveillance covers a wide variety of sources managed through a quality management system, so make sure this is integrated into your current processes or create new processes as required.
Save time and frustration! Download a fully linked IVDR Table of Contents that includes the original IVDR text.
Article 82 details requirements for reporting incidents. As with medical devices, the IVDR mandates that your reporting take into account the severity of the incident. You are required to report an incident no later than 15 calendar days after you become aware of it. Incidents that might pose serious public health threats must be reported within 2 calendar days. Thus, if you learn about something on Friday you better report it by Sunday, even if it’s a preliminary report that will be appended later – no exceptions. If the incident caused a death or “serious deterioration in a person’s state of health,” the reporting timeframe is 10 days. There are many additional requirements in Article 82, so you’ll want to study this carefully.
Many companies have been performing PMS activities all along, especially if they’re marketing in other countries. However, this may seem like a bureaucratic overreach for companies that have not been required to do so and whose products have excellent safety records. Alas, there is a logical reason for it. Following the implementation of the Medical Device Regulation (EU 2017/745) will come the rollout of the long-planned EUDAMED database. EUDAMED will also be used for IVDs, giving European Competent Authorities in all EU countries a centralized reference to track serious incidents, field safety notices, corrective actions, and Periodic Safety Update Reports (PSUR) – all in the name of greater transparency and patient safety.
If you have not already started preparing for your transition to the European In Vitro Diagnostic Regulation, here are some recommendations on where to start.
Conduct a robust assessment to see where you stand with regard to future compliance in Europe. A gap assessment is the first step and should answer these questions, among others: What classification will your IVDs have under the IVDR? What performance and clinical data do you already have to support registration, and is it robust enough? Do you have a quality management system in place? Are you prepared to get certified to ISO 13485:2016 if needed? An important first step is for you and your team to review the existing Essential Requirements found in the IVDD and compare those to the new General Safety and Performance Requirements (GSPR) found in the IVDR.
If you manufacture a range of IVD products, it is vital that you come up with a transition plan for those products that takes the following into account:
The IVDR will apply to 85-90% of IVDs sold in Europe, up from 10-15% now. You don’t have to be a math genius to figure out that demands for Notified Body services will rise exponentially. It is already very difficult to get Notified Body services for audits and product technical reviews. Each Notified Body must be designated to be allowed to certify IVDs and not all Notified Bodies are designated to handle all IVD categories. Likewise, reference labs are going to be swamped with testing of higher-classified products. You should start discussions with Notified Bodies and these labs right away to make sure they will be able to handle your specific products and ensure your place in line.
The IVDR will require input from a range of players involved in the manufacturing, importation, and sale of your products. These entities, who now fall under designation of Economic Operators (as defined in Article 2), will take on new responsibilities. As the manufacturer, it is your job to make sure that your EU Authorized Representative, importers, and distributors are all ready to assume their new obligations, which include adding information to the EUDAMED database. Set up meetings with your key distributors and importers and ask them about their plans to prepare for IVDR transition.
Even for experienced IVD regulatory professionals, theres a lot of information to digest in the IVDR. Its quite easy to miss something given the complexity of the regulation and its many cross-references. Part of your early planning process should include staff training. This could take the form of regularly scheduled internal meetings to discuss various aspects of the regulation or training on the IVDR. Either way, start early so everyone understands the requirements and the plan for meeting them. If you have numerous products in your portfolio, starting early becomes even more critical.
Plenty of companies selling in Europe and other countries with minimal regulatory oversight of IVDs will take one look at the IVDR and ultimately decide to throw in the towel. There are perfectly rational reasons to call it quits. Some products might be nearing the end of their useful life or might have such low margins or revenue that the additional cost of compliance exceeds the profitability of the products sold in the European market. These are discussions to have early on after you have conducted a gap analysis.
There is a glass half-full aspect to all of this. The IVDR presents an opportunity for IVD companies that don’t already sell in the US market. How? Companies that decide to make the extra effort to comply with the IVDR will find themselves in an excellent position to gain 510(k) clearance in the US should they choose to do so opening the door to another enormous market.
If there’s one good thing to come out of the IVDR, it’s that a whole lot of people with IVD regulatory experience can expect absolute job security for years to come. (That means you.) However, it also underscores the need to staff up earlier than you might otherwise plan to do. Don’t wait until mass panic ensues and you are competing with everyone else for talent. The issues of resources should not be ignored. You are not a superhero (yet) and your existing workload won’t disappear during IVDR transition time. Plan out the migration of products to the IVDR and try to estimate how much work will be required. Figure out what your existing team will realistically be able to handle, how much staff you will need to add, and what you want to outsource. If you plan to work with outside consultants, establish a relationship early on to help you plan because they, too, will have more work than they can handle as the deadline gets closer. Surprise, we can help with that.
If you really want to take a deep dive into the IVDR, consider our in-depth training class on the IVDR. We can also assist with IVDR consulting.

US OfficeWashington DC
EU OfficeCork, Ireland


