
Medical device manufacturers and regulators generally have different goals, but one thing they have in common is a desire to make sure devices are safe and effective. To achieve that, regulatory managers often spend a substantial amount of time analyzing, evaluating, and controlling all manner of risks.
Residual risk analysis takes individual risks that meet your acceptance criteria and examines the risk they pose individually as well as all risks in aggregate. It is entirely possible that individual risks might meet your risk acceptance criteria on their own but not when evaluated in aggregate. An example of this might be cybersecurity threats for an insulin pump. Individual vulnerabilities might be analyzed, evaluated, and mitigated as far as possible. While some risks remain, they are determined to be acceptable. Collectively, those small vulnerabilities may allow a hacker easier access to penetrate the software than initially thought and control the device, causing harm to the patient.
Medical device and in vitro diagnostic (IVD) manufacturers typically take one of two approaches to risk acceptability.
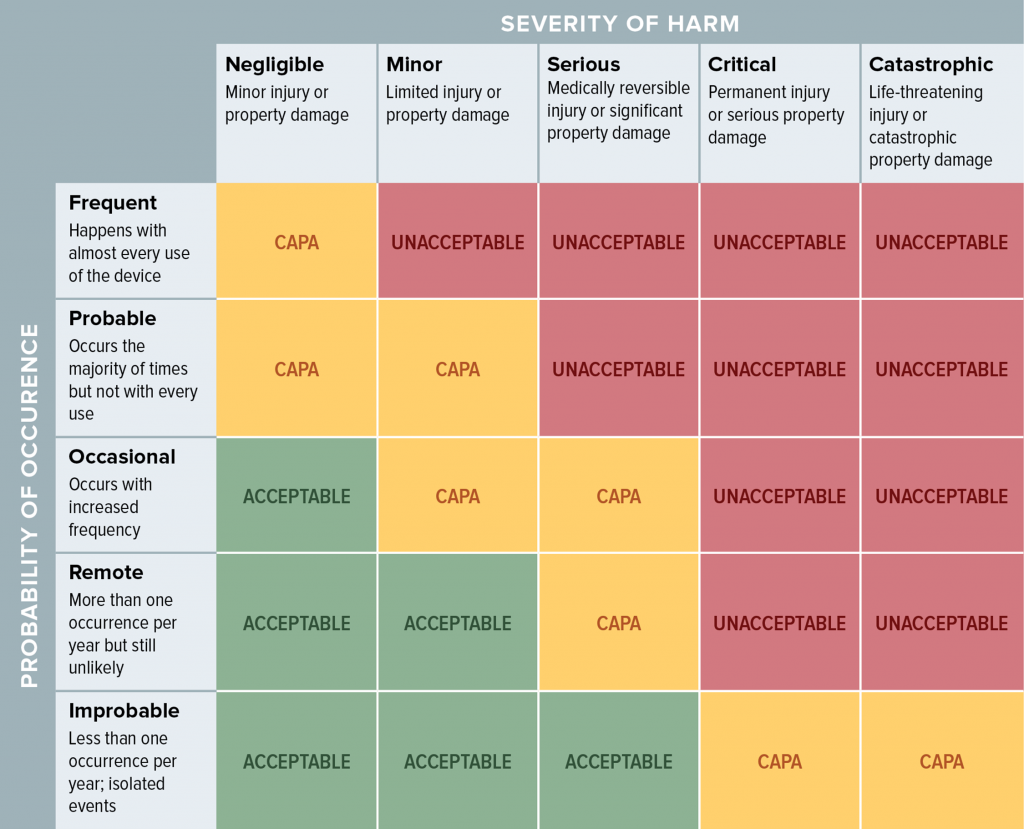
When evaluating risk, many people use a table such as this to look at the probability of occurrence and severity of harm. With residual risk, you can consider additional factors, such as the benefit the product offers when used as intended.
There is no magic formula for determining how much risk is acceptable, and you should avoid coming up with numerical point systems as a means of making this determination. As previously stated, while the criteria for measuring the residual risk may be the same as the initial evaluation, the criteria for determining the acceptability of the risk should be based on the benefit of the product to the patient. It is up to you to establish a method of evaluation, but you should involve people who have the knowledge, experience (medical and/or clinical knowledge), and authority to make this determination. The results of your evaluation will become a part of your risk management file.
More Diversity = Better Results
When we talk about diversity, we are talking about assembling a team that covers the gamut of manufacturing, distribution, customer service, sales, packaging, and so on. For instance, without a colleague from manufacturing, nobody will be able to foresee potential manufacturing problems that could create hazardous situations. Without someone from customer service, you may not get insight into how consumers might use your device outside of its indications for use despite your well-labeled warnings and magnificent instructions for use. Residual risk is often an exercise in brainstorming, and you will get far more ideas on what can go wrong with a team that represents many areas of your company beyond RA/QA.
If you want to take a deep dive into ISO 14971, consider the Oriel STAT A MATRIX risk management training course. Our team is also available to help you comply with EU MDR or FDA risk management requirements.

US OfficeWashington DC
EU OfficeCork, Ireland


