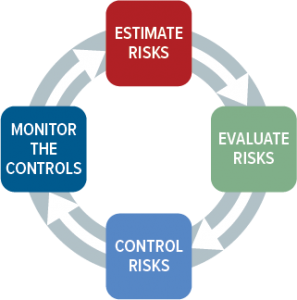
From a distance, risk management seems straightforward. You have a device, evaluate its potential risks, mitigate those risks, monitor them over time, and you are done. Seems easy, right? Ah, if only life were so straightforward. The reality is that risk management is one of the more complex aspects of regulatory compliance, simply because risk comes in so many flavors and perceptions of severity, and probability can be interpreted quite differently.
The thing that makes risk management tricky is that we often don’t have enough real-world data to accurately quantify risks, especially for new devices. Fortunately, there is a systematic process you can establish to estimate, evaluate, control, and monitor risks. Before we get into that, let’s step back and talk about the regulations and standards that dictate how you should approach risk management.
Simply put, we have a collective interest in ensuring that medical devices are safe and effective. Risk management is not optional it is a regulatory requirement worldwide. The US FDA mandates it in the Quality System Regulation (21 CFR Part 820). Europe requires it in the new Medical Device Regulation (MDR 2017/745). Likewise, Japan, Canada, Australia, Brazil, and all other major markets require the application of risk management, which is either referenced in their national regulations and/or ISO 13485:2016.
Fortunately, national governments have NOT created their own guidelines telling you how to how to perform risk management. Instead, they all defer to ISO 14971, the global standard for medical device risk management. The intent of the standard is to identify hazards associated with medical devices at all stages in its life cycle, from product design to procurement to production and postmarket use. In all cases, the goal is to estimate, evaluate, control, and monitor the risks associated with each life-cycle stage. There are two versions of this standard in use today:
If you are just getting started implementing risk management for your company, purchase the ISO 14971:2012 standard and its guidance ISO 24971:2013. You will also want to buy and read the ISO/TR 24971:2013 standard. It is brief but provides excellent guidance for dealing with specific areas of ISO 14971. Both are copyrighted documents and you can purchase them online from ISO.
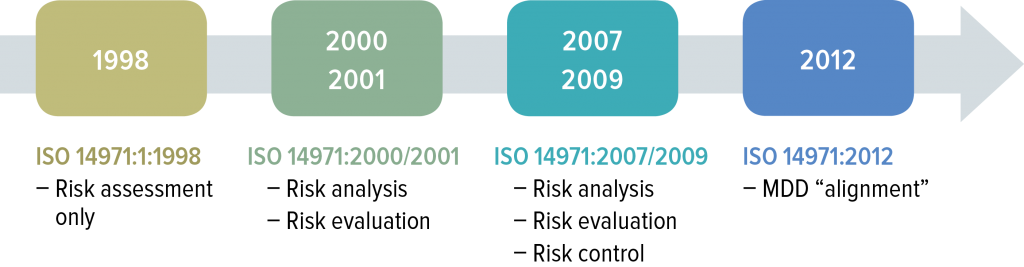
ISO 14971 was first introduced in 1998 and has expanded in scope during subsequent releases. Work on an updated version is underway.
An updated dated ISO 14971 is underway and expected to be complete sometime in 2019. The focus of the revision is not on revising the risk management process but rather to improve the information on implementation of the life cycle risk management process.
Although risk management can be complex, the main body of the ISO 14971 standard is a scant 14 pages and consists of 9 clauses:
And these are the key annexes supporting those clauses:
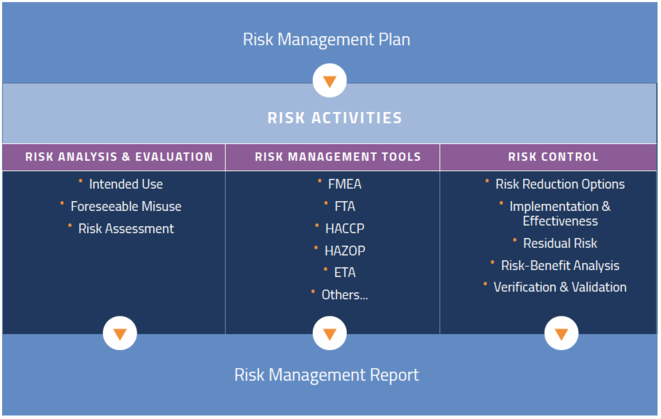
So where to begin? It helps to think about risk management as a process that starts with a plan. While the end deliverable is a report, your work in controlling risk is never done. We will talk in detail about each of these areas later, but here are the steps.
If you enjoyed this article, be sure to read the second post in this series focusing on risk management planning. If you’re ready to take the next step, check out our intensive risk management training class.

US OfficeWashington DC
EU OfficeCork, Ireland


