QA/RA Consulting, Auditing & Training

Article contents:
Think of Design Control as a Framework
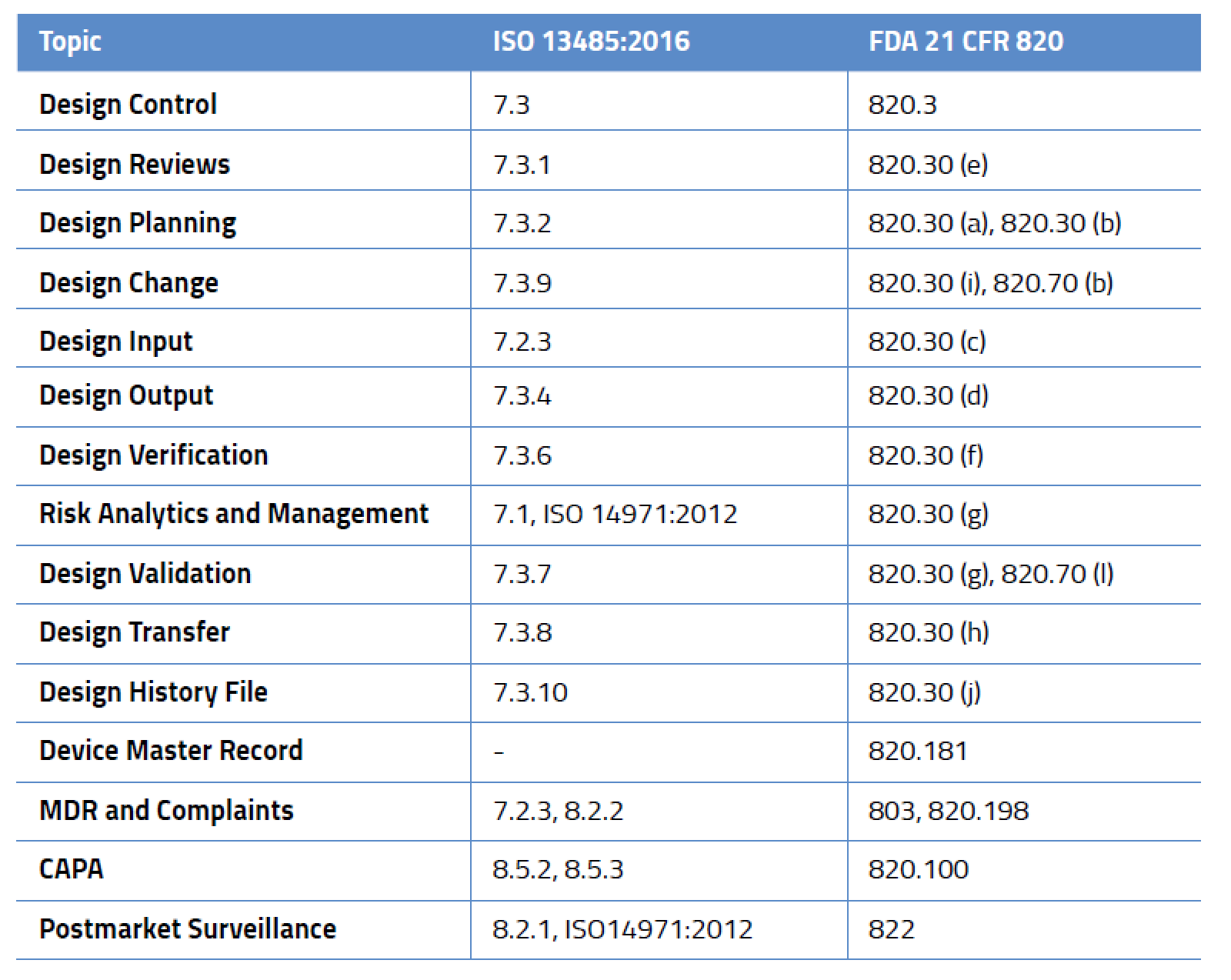
Design Control-Related Processes in ISO 13485:2016 and FDA 21 CFR 820
When Does Design Control Begin and End?
The Overall Design Control Model
Setting the Stage with Design Reviews
Medical Device Design Control Planning
Change and Configuration Management
Medical Device Design Inputs, Outputs, Verification, and Validation
Medical Device Design Outputs
Verifying the Final Medical Device Design
What’s the Difference Between Medical Device Verification and Validation?
Medical Device Design Validation
What’s the Difference Between a DHF, DMR, and DHR?
What Is a Medical Device Design History File (DHF)?
What Is a Device Master Record (DMR)?
What Is a Device History Record (DHR)?
Coding changes. Materials substitution. Design refinements. UX improvements. Manufacturing changes. Medical devices, like all products and software, are constantly evolving. Yet, due to the nature of medical devices and their impact on personal health and safety, the entire product life cycle of a device must be carefully documented. It’s an easy thing to overlook. Developers and engineers get so immersed in the “design and refine” process that they sometimes forget about their obligation to document how they got from A to B.
This documentation process is widely known as “design controls” and its purpose is to ensure that medical devices meet user needs, intended uses, and specified requirements. Design controls apply to the design of the product and associated manufacturing processes. The US FDA has identified lack of design controls as one of the major causes of device recalls, and therefore they want you to pay specific attention to the following:
While design control is not required for all medical devices, it applies to nearly every medium- and high-risk device. And this isn’t just for new devices – in the US, FDA requires design controls for all Class II and III medical devices and even some Class I devices (notable among these are Class I devices automated with software). Likewise, ISO 13485:2016 mandates design controls. It is important to remember that design control requirements are a framework and not a prescriptive solution. Regulators set the expectations but don’t give you the answers. It’s entirely up to you to define your processes, defend them as appropriate, and prove they meet the requirements outlined in the table below.
It never ends. Design control is not a “once and done” process – it applies to modifications or improvements to existing designs, or changes to processes. Design control does not, however, apply to the ideation stage of medical device development. You don’t need to document the development of prototype concepts or feasibility studies. However, you do need to start creating a plan once you have decided that a specific design will be developed. To assist regulators, you should document the flow of the design process so it is very clear where research is ending and development of the chosen design is beginning.
Even if a device has been in the marketplace for years, design control is never finished. Both large and small changes to a device or associated manufacturing process must be evaluated for their impact on product performance, safety, and use.
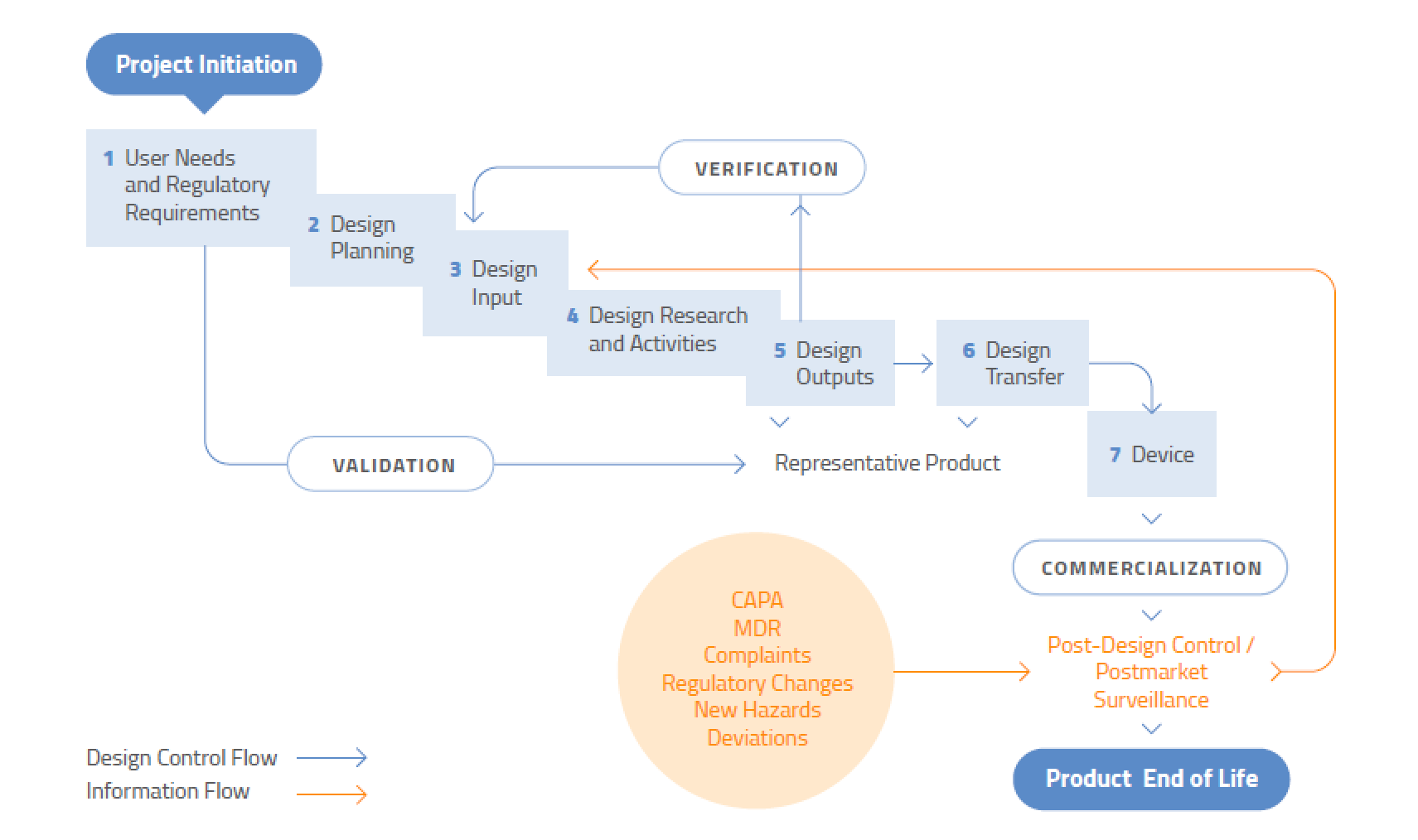
Source: US FDA
Design reviews are an important part of the overall design process. The objective is to establish a process to make sure the design you created (the output) meets your design requirements (the input). A design review provides a mechanism that confirms the design is ready to move to the next stage of the process. Although it is technically focused, a design review is not limited to only the design of the product – the resulting analysis provides feedback for the design team. Both FDA and ISO 13485:2016 require you to conduct and document these formal reviews on a regular basis at appropriate stages during the development process. The documented results become a part of the DHF. Those reviews must include representatives from all functions involved in the design process and any other specialist personnel. FDA also requires you to add an independent reviewer without direct responsibility for the design stage being assessed.
The number and types of reviews are project-dependent, so define them in the plan. Review frequency will be impacted by the size and scope of the project and the product development life cycle. At minimum, two design reviews should be held – one at the beginning during the design inputs review process and another at the end during the design transfer phase.
Before you can control your product design, you need a plan for doing so. Design control planning enables the management team to exert more control over the R&D process by clearly communicating policies, procedures, and goals to the development team. It also provides a basis for measuring conformance to quality system objectives. This means you must:
The form and organization of the planning documents are less important than their content and should be customized for each company. Remember that as development progresses, the planning documents should be updated.
Product development is inherently an evolutionary process. While change is a healthy and necessary part of product development, quality can be maintained only if those changes are controlled and documented. As such, a key component of design control involves establishing procedures to identify, document, review, verify, validate, and approve design changes before they are implemented. You are not expected to maintain records of all changes proposed during the very early stages of the design process; however, all design changes made after the first design review must be documented. These records create a history of the evolution of the design, which can be useful for failure investigation and for facilitating the design of similar products in the future. Such records can also prevent repeat errors and the development of unsafe or ineffective designs.
Examples of design changes that must be controlled include:
While you are required to document changes, the extent of your evaluation and documentation should be in direct proportion to the significance of the change and its effect on other parts and product in process or already delivered. You’ll also need to establish a system of traceability for this documentation, a process called “configuration management.” This process involves identifying the documentation to be controlled, the procedures for controlling the documentation, and the responsibilities of those managing the documentation.
Of all the design control activities, developing a solid foundation of requirements is the most important. Why? The requirements that make up the design inputs establish a basis for performing subsequent design tasks and validating the design. Section 7.3.3 of ISO 13485:2016 and section 820.30 of the FDA QSR talk about design inputs. Essentially, the key requirements include a need to determine:
All of these inputs must be documented, reviewed, and approved for adequacy, and you need to make sure that the requirements are complete, clear, and don’t conflict with one another. Basically, design inputs should specify what the design is intended to do but they should avoid specific design solutions. Your product developers play a key role (but not the only one) in developing the design input requirements, regardless of who developed the initial product concept. They ultimately bear responsibility for translating needs into a set of requirements that can be validated prior to implementation.
As you define inputs, it is important that you properly identify the customers using your device. Are they lay consumers? Doctors? Medical technicians? This is vital, because all design verifications and validations are matched to customer needs and related technical requirements. Also, while your developers will play a key role in developing inputs, you should also get input from people closest to the customer, such as people in marketing, tech support, and sales. For complex designs, it is not uncommon for this design input stage to consume as much as 30% of the total project time.
Design control is not just about controlling the design inputs. It is also essential to know whether those design and development inputs result in the right outputs – namely, whether the design you created is safe and functions as planned. Design output requirements are intended to apply to all stages of the design process, and to characterize important aspects of the design.
Design output documents must define the critical outputs (specifications, properties) and reference your acceptance criteria. All of this must be documented, reviewed, and approved before the design is released. The outputs also provide needed important information for purchasing, production, and servicing. Of course, this begs the question: What constitutes design output? Here are some examples:
Again, all of these outputs must be verified against applicable design inputs. The finished design output documentation is the basis for your device master record (DMR).
Risk management is a critical part of regulatory compliance and has a direct link to design control. It is the systematic application of management policies, procedures, and practices to the tasks of identifying, analyzing, controlling, and monitoring risk. It should be used early and throughout the design and development process and often generates new information that needs to be fed back into the design and development process.
In simple terms, design verification proves that you built the device correctly according to all specifications. During this process, you will ensure that all of your design outputs meet the design inputs. You need to establish a procedure for verifying the device design and maintain records of the results of the verification and any subsequent actions. Verification can be accomplished using one or more of the following:
Your design verification records need to include the date and person who performed the verification as well as methods, acceptance criteria, and any statistical techniques used. This all becomes part of your design history file (DHF). The US FDA likes to see these activities displayed in a traceability matrix.
Don’t conflate verification testing with production testing. Verification testing proves conformance with documented design outputs and inputs, whereas production testing determines whether the unit being tested has been manufactured correctly. Production testing is rarely comprehensive enough to verify the design.
In simple terms, verification challenges the design throughout its development to see if you designed the device correctly. Validation is a summation of those efforts to ensure that your design meets user needs and intended uses. In other words: You verify that you designed the device according to spec and you validate that it meets the needs of your users.
There are two common types of validation. Process validation uses objective evidence to make sure a process consistently produces the same result. Design validation, on the other hand, ensures that the device meets user needs and intended uses and will therefore become a viable product in the marketplace.
FDA requires you to perform design validation “under defined operating conditions on initial production units, lots, batches or their equivalents.” This includes software. What does “defined operating conditions” mean? Basically, you need to ensure that the conditions under which the product is made for validation purposes are the same conditions anticipated for full-scale production.
As with all other aspects of design control, you need to establish a procedure for validating your design. Your validation activities should address the needs of all stakeholders – including patients, healthcare workers, installers/servicers – and the physical healthcare environment itself. Your validation needs to be performed based on each intended use.
As for timing, the design validation is completed after clinical evaluation is complete. Your clinical evaluation may include a clinical trial but is more likely to consist of safety testing, literature searches, etc. Also, regulators know that it is not always practical to perform clinical studies on finished products, so you are allowed to use prototypes. However, final design validation must be conducted on actual production devices.
A properly executed validation of design may end up poking holes in your original assumptions, and for this reason you may want to have the validation performed by an independent team familiar with the details of the project but not directly involved in the design. All of your validation activities become part of your design history file (DHF).
Congratulations! You’ve created all of your design inputs, defined the outputs, and conducted design verification. Your design is complete and you are ready for production. Now it’s time to make sure those design outputs are correctly translated into written production specifications. These specifications are typically composed of written documents, including:
In some cases, the design transfer may come before validation when your ability to produce a prototype or production-level device cannot be achieved in another manner. If that’s the case, you will need to perform another design review during the design transfer process and create a cross-functional team that includes representatives from engineering, manufacturing, materials management, quality, and business development. A design transfer checklist will help you ensure that nothing is forgotten.
These records, while confusingly similar in name, are quite different in purpose and are cornerstones of the design control process. It is vitally important you don’t conflate them. Before we talk about each of them, here is a quick primer on how they differ.

As the name implies, the design history file is your repository for all records that demonstrate your medical device was developed in accordance with the approved design plan. You are required to maintain a DHF for each type of device. The DHF contains or references:
There are no specific requirements saying how you must organize your design history file or where you must store it. For simple products, the design engineer may assemble and maintain the entire DHF. However, for larger projects, you will most likely need a document control system housed in a centralized location. Your DHF and associated documents are an important part of any US FDA 510(k) or PMA submission.
The DHF shows how you developed your recipe, but the DMR is the recipe itself. In other words, it contains all the information needed to produce the device. Section 820.181 of the FDA QSR is specific about what the device master record should contain, including:
In maintaining this information, it is most important that you think of the DMR as the place where you can store files and link to other data sources rather than keeping everything in the DMR itself. ISO 13485:2016 combines the DHF, DMR, and other documentation into a file known as the medical device file.
The device history record (DHR) is demonstrable proof that you followed the recipe, namely your DMR. It is required by FDA, but ISO 13485:2016 contains no such requirement. FDA specifies that your DHR must include the date of manufacturing for each batch, lot, or unit; the number you manufactured; the quantity released for distribution; the labeling used for each production unit; plus any UDI, UPC, or other identification used. You must also include your acceptance records, which show that you followed the recipe. Your DHR is particularly important during audits because the investigator may want to see your DHR and compare it against your DMR for compliance. If you are using a contract manufacturer, make sure your contract and device history record specify who reviews and releases the device.
If you enjoyed this article, check out our intensive medical device design control training class. We also offer a design control class for combination products. Also, if you’re not sure where to start with implementing design controls in your company, we can help.

US OfficeWashington DC
EU OfficeCork, Ireland


