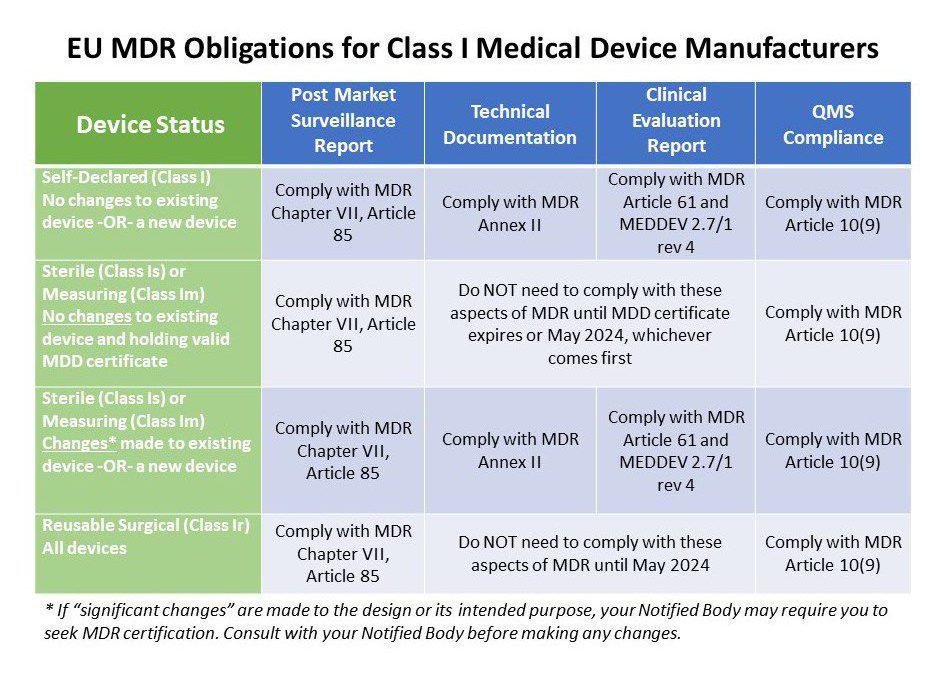
1 – Post Market Surveillance Reports (PMSR) for Class I devices
Even if you are not planning to make any changes to your Class I device anytime soon and will continue to declare conformity with the MDD, ALL Class I manufacturers are required to generate a PMS plan and PMSR as outlined in Chapter VII, Article 85 of the EU MDR. Your PMSR needs to summarize the results and conclusions of your PMS data along with a rationale and description of any corrective actions taken for products on the market. This report becomes part of your technical documentation and is updated when necessary and made available to EU Competent Authorities upon request. Your PMSR should be maintained throughout the entire life cycle of the device.
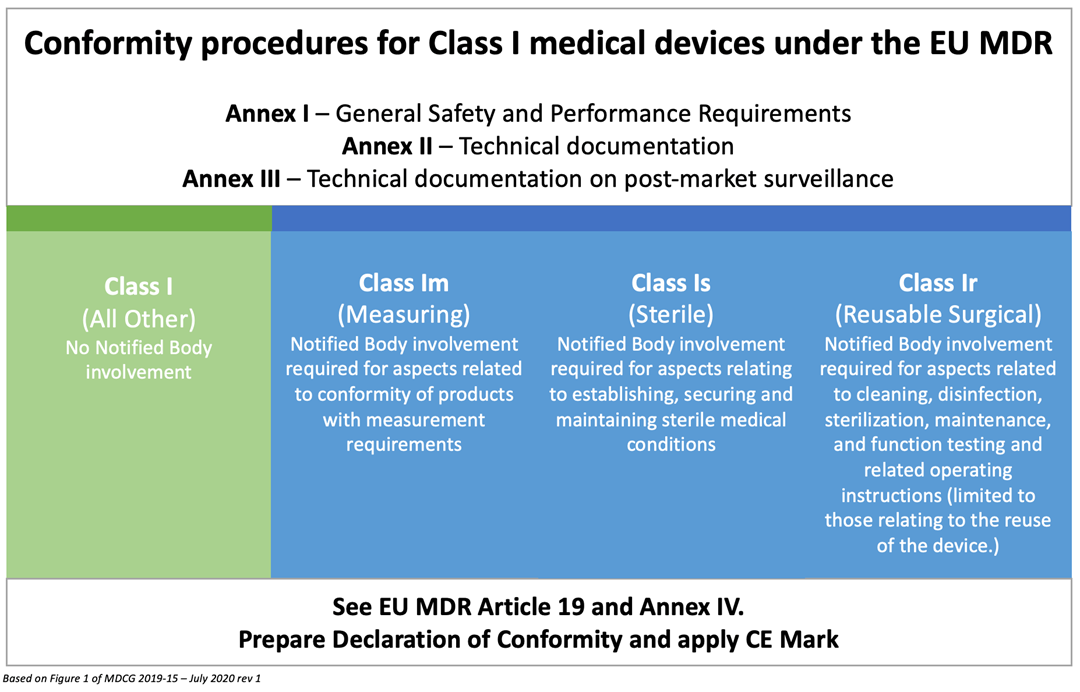
2 – Technical Documentation for Class I devices
Here’s where things get a little trickier. The table above explains the MDR obligations of various Class I device manufacturers. Annex II of the MDR requires all medical device manufacturers to have adequate technical documentation. This includes very detailed information on the device, previous and similar generations of the device, a complete set of labels for the device, design and manufacturing information, general safety and performance requirements, a benefit risk analysis and risk management information, product verification and validation data, clinical data and more.
The Corrigendum we mentioned earlier does give a free pass to Class I devices that are provided sterile or have a measuring function if no changes are made and they are still holding a valid MDD certificate. That’s true for all reusable surgical instruments as well, which were previously self-certified. However, if you are marketing any new Class I device, or you are making changes to the design or intended use, you must comply with Annex II of the MDR. Study it carefully.
3 – Clinical Evaluation Reports (CER) for Class I devices
If you manufacture Class I devices that are provided sterile or have a measuring function you may already have a technical file in compliance with the MDD. However, your old technical file will not stand up to the strict new requirements of Chapter VI of the MDR and MEDDEV 2.7/1 rev. 4. Again, the situation is a little tricky because the same situation we just described for technical documentation applies for clinical data as well.
What’s different is that clinical data takes time to collect and you can’t simply postpone collecting the data necessary to comply with the new requirements until your existing MDD certificate is about to expire. You’ll be in a world of hurt if you do.
Whatever your situation, we recommend that you start with Section 7 (pp 15-17) of the MEDDEV as it discusses the scope of the clinical evaluation needed. This will be critically important for you from now on. You can roll your eyes all you want but a Notified Body will not care if your device has been safely sold for 25 years – you still need to gather clinical evidence to demonstrate that your device is safe. Start now. This also provides an excellent overview of your CER obligations. If you are still thoroughly confused after reading all that, consider this CER class.
4 – Quality Management System for Class I manufacturers
If your company manufactures Class I sterile or measuring devices, you may already have a quality management system. If you have not already implemented a quality system that complies or is certified to ISO 13485:2016 you may want to consider doing so. We can help. While the MDR does not explicitly require a certified Quality Management System (QMS), the easiest way to comply with the QMS requirements in Article 10 is by achieving ISO 13485:2016 certification. Granted, you are not making pacemakers so your QMS can be proportionate to the risk of your device. Even self-certified Class I device manufacturers need to implement a basic QMS that includes procedures for management review, corrective action, vigilance, PMS, and so on. Read MDR Article 10 thoroughly. Starting from scratch? This class provides a focused deep dive primer on QMS compliance.