QA/RA Consulting, Auditing & Training
In this article:
Regulations Governing a Medical Device QMS
Relationship Between Medical Device Approval and Your QMS
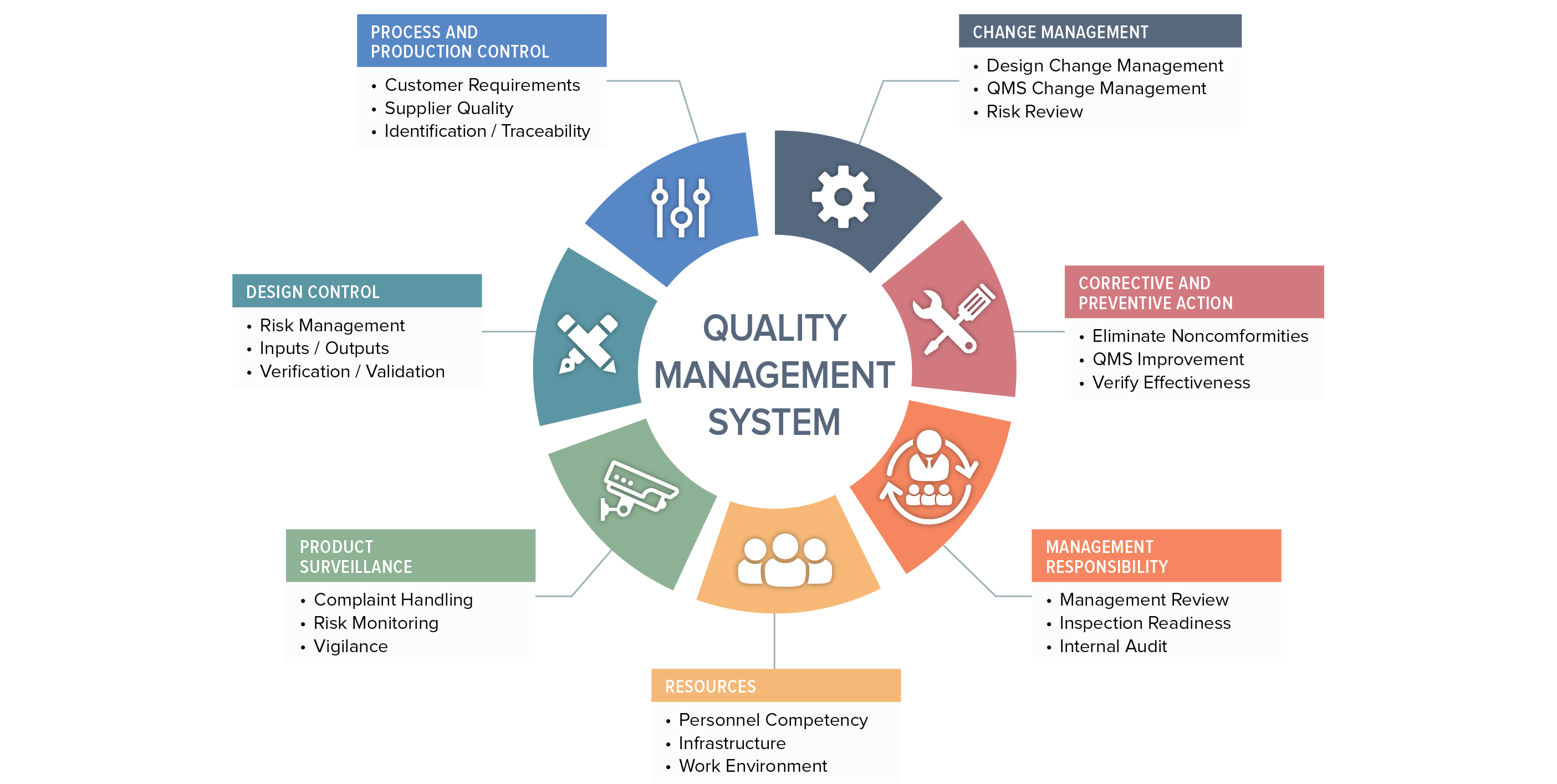
Key Components of a Quality Management System that Meets US FDA and EU Requirements
Developing a Medical Device QMS
Understanding Key Obligations in ISO 13485:2016 and the US FDA QSR
Let’s start with the basics. In simple terms, a medical device quality management system (QMS) is a structured system of procedures and processes covering all aspects of design, manufacturing, supplier management, risk management, complaint handling, clinical data, storage, distribution, product labeling, and more. Most medical devices will require some form of a QMS; the complexity of the QMS will vary based on the classification of the device. For example, companies making medium-risk (Class II) or high-risk devices (Class III) devices will require a different QMS implementation than companies making low-risk devices (Class I). We won’t get into the specifics of product registration or classification, but you will want to understand the classification of your devices before building a QMS.
Nearly every major market requires the implementation and maintenance of a quality management system as a condition of product registration. Device manufacturers in Europe tend to follow the ISO 13485 standard, while US companies comply with the US FDAs Quality System Regulation (QSR).
ISO 13485 is an international quality management system standard followed by companies selling in Europe, Canada, Australia, and other markets. Except for Canada, application of ISO 13485 is not actually required, but it is the de facto means by which most companies comply with the specific QMS requirements set forth in national medical device regulations.
The US has its own set of regulations for medical device companies. The US FDA QSR, also known by its US regulation number 21 CFR Part 820, preceded the original publication of ISO 13485. US medical device companies that distribute their products internationally need to meet the requirements of both. Similarly, countries outside the US that distribute products in the US must also comply with US FDA 21 CFR Part 820.
Other countries such as Brazil and Japan have their own nuanced QMS requirements, but those are based on ISO 13485 or the FDA QSR. The good news is that many of the requirements of ISO 13485 and the FDA QSR are very similar. As such, companies can have a single, harmonized quality management system that meets US, Canadian, European, and any other regulatory QMS requirements. Harmonizing disparate quality management systems into one integrated system may seem daunting but in the long run the effort is well worth it. We will talk about the requirements of ISO 13485 and FDA QSR in more detail later.
You may wonder what a QMS has to do with getting approval for your product(s) from the US FDA or obtaining CE Marking in the European Union. They are inextricably linked. The US FDA requires compliance with 21 CFR Part 820 at the time your product is registered with FDA. The majority of companies making medium-risk devices will go through the 510(k) process. When you submit your 510(k), you are expected to be in compliance with 21 CFR Part 820. Ironically, although QSR compliance is required, FDA does not require proof of compliance when registering your Class I or Class II device. Why? The US FDA enforces compliance through random inspections. As such, FDA inspectors may come knocking on the door of the manufacturer of a newly registered medical device at any time. If you are not fully prepared, you won’t like the consequences.
It’s a different process in most European countries, where you need to obtain CE Marking for the device as a condition of distribution. If you are seeking to obtain CE Marking for anything other than a Class I non-sterile, non-measuring, non-reusable surgical instrument device, you cannot get CE Marking without proving you meet the requirements of Article 10 (General obligations of manufacturers) and Annex IX (Conformity assessment based on QMS and technical documentation) of the new EU Medical Device Regulations (EU MDR). The most common way companies meet the requirements of Article 10 and Annex IX is through third-party certification to ISO 13485. Third-party certification is conducted by auditing organizations known as Notified Bodies (NBs). We should note that there are other ways to meet European QMS requirements, including the requirements listed in Annexes X (Conformity assessment based on type examination) and XI (Conformity assessment based on product conformity verification).
Finally, Health Canada (the Ministry of Health) requires conformance to ISO 13485:2016 through MDSAP certification. If you have plans to sell in Canada, you will need to implement a QMS compliant with ISO 13485:2016.
We’ve established that, as a medical device company, you need to implement a QMS and be in compliance with FDA 21 CFR Part 820 and/or ISO 13485:2016. As noted, ISO 13485 is not required in most parts of the world but certainly is the “de facto” means of meeting QMS requirements in many countries. Let’s talk about the entities with a role in maintaining, auditing, and certifying quality systems, starting with the US.
Within the Food and Drug Administration (FDA) is the Center for Devices and Radiological Health. This division – commonly known as CDRH – oversees the regulation of most medical devices that do not have a pharmaceutical or biological component. When you register your company with the US FDA (establishment registration usually is done when you also register your first device), you will be on the “radar” of CDRH inspectors. As mentioned earlier, the FDA conducts its own inspections and holds the legal authority to prevent or stop companies from selling their products in the US market.
European Notified Bodies are supervised by the Competent Authorities of a particular EU member state to conduct inspections of medical device manufacturers. You’ll find a list of notified bodies which can assess the conformity of medical device products listed in the NANDO database. Even though Notified Bodies are not government agencies, they do have the power to grant or deny ISO certification and/or CE Marking for your device. In Canada, these are called Registrars, but nearly all Registrars are also Notified Bodies. Under the MDSAP scheme, Registrars are referred to as Auditing Organizations.
If you are studying medical device quality management system compliance, you also might run across the terms Authorized Representative (Europe), Sponsor (Australia), D-MAH (Japan), and US Agent. If you are a US company selling in Europe, for example, and you don’t have a local office there, you need to appoint a regulatory representative in that country (or countries). Regulatory representatives don’t really play a role in the management or inspection of your quality system, but Notified Bodies will most certainly check to make sure you have one during their audits.
If you are wondering where to start in putting together a quality management system for your organization, here are the basic phases.
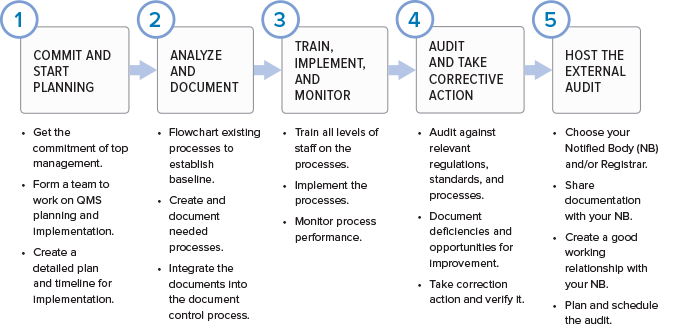
While regulatory imperative is the driving force behind the creation of a quality management system, the QMS is not a set of procedures that get stored on a hard drive only to be opened when inspectors arrive. If you are doing things correctly, your QMS should help your business to be successful and evolve over time. Processes will be added, refined, or eliminated. The QMS should be built using a process approach (thus enabling the organization to plan processes and their interactions) and incorporate a risk-based approach and the PDCA cycle.
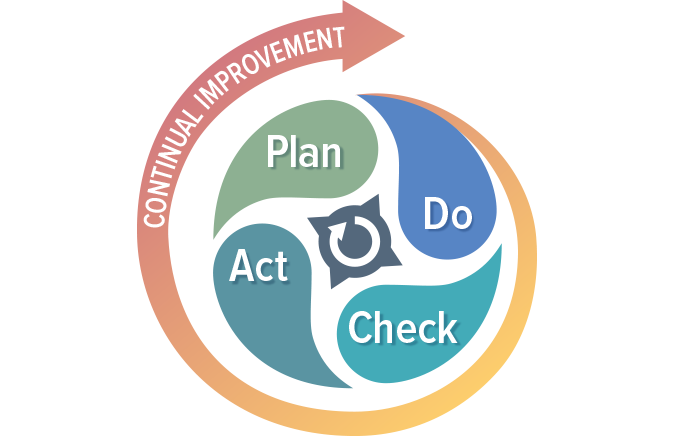
The PDCA cycle enables your organization to ensure that its processes are resourced adequately, managed in practice, and analyzed for improvement opportunities that may be acted upon. Coupling a risk-based approach with your process management activities enables your organization to determine the factors that could cause your processes and QMS to deviate from the planned results. Coupling these two approaches will help your organization put preventive measures in place to minimize negative outcomes.
We can’t cover all aspects of the ISO 13485:20216 standard and 21 CFR Part 820 regulation in this discussion, so we will focus on five key areas:
A QMS cannot function without solid document control. Section 4.2 and other specific sections of ISO 13485:2016 outline your obligations, and so do various subparts of the FDA QSR, as indicated below. There are several important documents you must maintain – this is only a partial list:
You may have excellent control over your records, but without the full support of executive management your QMS will not be effective at maintaining product safety and promoting continuous improvement of your processes. You’ll notice that we used the word “executive.” FDA and the ISO standard are serious about this. FDA states in 820.12, “Management with executive responsibility shall establish its policy and objectives for, and commitment to, quality. Management with executive responsibility shall ensure that the quality policy is understood, implemented, and maintained at all levels of the organization.”
The primary difference between the QSR and ISO 13485 is that the standard emphasizes management responsibility in the context of meeting customer requirements in addition to regulatory requirements. The QSR is focused entirely on meeting requirements on design, manufacturing, distribution, and support of safe and effective medical devices. You will find the requirements for management responsibility in section 5 of ISO 13485:2016.
Section 6 of ISO 13485:2016 and subparts 820.20/25/70 deal with this topic, and touch on a variety of issues. Essentially, they require the company to identify the need for and to allocate qualified personnel, overall infrastructure, and work environment to ensure product safety. You also have an obligation to ensure the competency of your staff, which includes establishing formal competency and training procedures, maintaining records related to employee competency, and providing training as needed. Don’t take these requirements lightly. FDA often finds that failure to comply with these subparts of the regulation leads to other significant regulatory violations.
Section 7 of ISO 13485:2016 is important, along with corresponding subparts of the FDA QSR. One of the more important areas covered here concerns your obligations to maintain design controls. While the objective of maintaining control over your product design (which includes software) is consistent between the ISO standard and the QSR, the standard applies to all medical devices whereas the QSR requires a design history file primarily for medium- and high-risk devices only.
Purchasing controls (21 CFR Part 820.50) are covered under product realization. This includes a requirement to document supplier controls, subcontractors, purchasing data, receiving, etc. The focus here is on ensuring that purchased product doesn’t have a negative effect on the quality of your medical device.
Section 7.1 specifically requires the use of risk management in product realization and references a related standard, ISO 14971. If you are not already familiar with this standard, you’ll need to be. Risk management is hugely important and is critical to ensuring a safe and effective device. The new European Medical Device Regulation also places much more emphasis on this topic.
Section 7.5 of ISO 13485:2016 on production and service provision covers topics including the acceptance criteria for products and parts, contamination control, installation, product servicing, and even particular requirements for sterile medical devices – all of the activities that you actually perform to make or deliver your device. Read 21 CFR Parts 820.70, 820.80, 820.170, and 820.200 for specific information on FDA requirements for these areas.
The topic of process validation could be its own book, but at a high level, here’s what you need to know. Process validation is the process by which you establish objective evidence that a process consistently produces a result or product that meets predetermined specifications. Section 7.5.6 of the standard outlines the requirements along with 21 CFR Part 820.75. Validation requirements also apply to software you use throughout your processes.
Identification (section 7.5.8) and traceability (section 7.5.9) are also critical subsections of the standard and correlate with several sections of the QSR, including 820.60 and 820.65, respectively. The emphasis here is on making sure you know where your devices/components came from, how they were distributed, and where they are now. This issue has become far more important in the last five years. The US FDA has specific requirements for unique device identification (UDI). The European Union soon will have similar requirements for device tracking, information that will be added to the new EUDAMED database which requires close monitoring for the official implementation date.
Of course, a quality management system would be worthless without an effective means of measuring its performance in meeting planned results.
Section 8 of 13485 covers this topic and outlines your responsibility to collect feedback on your QMS from various sources, properly handle customer complaints, conduct regular internal audits, deal with nonconforming products, analyze data, and establish an effective corrective and preventive action (CAPA) program. You’ll find several sections of the QSR that take on these
Establishing a quality management system and understanding the differences between ISO 13485:2016 and the FDA Quality System Regulation can seem daunting. However, once you start to understand the principles behind the standard and the regulation, you will find plenty of overlap. We offer several ways to get started. Consider our medical device QMS overview training. If you want help building a QMS from scratch, we can assist with QMS implementation as well.

US OfficeWashington DC
EU OfficeCork, Ireland


