
The entire purpose of conducting validation is to build quality, safety, and effectiveness into the finished pharmaceutical, and not to rely on post-production batch testing. To accomplish this, you need to make sure that every process step performs as expected based on quality design, with the ultimate goal being consistent reproducibility. This process of validation is ongoing and conducted throughout the life cycle of the product, from development through commercial manufacturing.
Validation is not something you start and then figure out as you go; it’s complicated, and starting the process without a solid plan would be like wandering into the wilderness without a compass, water, or food. A validation master plan shows the interrelationships among processes and helps you plan resources and estimate costs, including those for external resources. A detailed VMP includes comprehensive lists of manufacturing equipment, testing equipment, special requirements, required documents, and more. Among other things, it also ties together all protocols necessary to complete the validation project and should clearly communicate the company’s intent to start production using the new process or equipment in a fully compliant state. It is important that senior management approves the validation plan so that they fully understand the scope of the effort before work begins. Creating a VMP checklist is a smart way to begin.
1. Design qualification (DQ) – Documented verification that the proposed design of the facilities, equipment, or systems is suitable for the intended purpose.
2. Installation qualification (IQ) – Documented verification that the equipment or systems, as installed or modified, comply with the approved design, the manufacturer’s recommendations, and/or user requirements.
3. Operational qualification (OQ) – Documented verification that the equipment or systems, as installed or modified, perform as intended throughout the anticipated operating ranges.
4. Process performance qualification (PQ) – Documented verification that the equipment and ancillary systems, as connected together, can perform effectively and reproducibly based on the approved process method and specifications.
Process validation begins by designing and/or understanding the commercial process used to manufacture your product. During this stage, your goal is to clearly define a process that is suitable for routine commercial manufacturing, delivers a product consistently, and meets its quality attributes. This stage will inevitably require you to run experiments and simulations. Regardless of what types of simulations you run, you must understand the degree to which your models represent the commercial process and try to predict the performance of it. Any protocols you conduct should use solid scientific methods and be well documented.
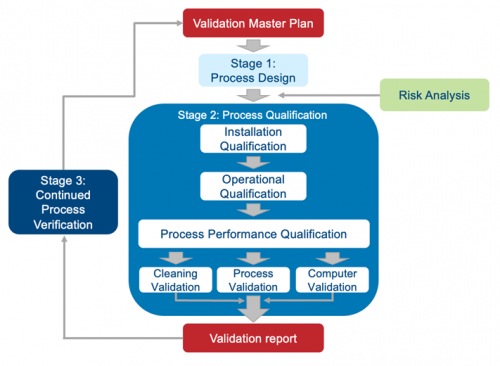
| Stage 1: Process Design Based on knowledge gained during development and scale-up activities, the commercial process is established. | Stage 2: Process Qualification Process design is evaluated to determine if it is capable of commercial manufacturing that is reproducible. |
There are a host of documents you need to gather in order to get a better understanding of what’s required for pharmaceutical process validation. Below we’ve listed the most commonly applied US and European regulations and guidance, plus international guidelines you will need to follow.
| US Food and Drug Administration (FDA) | European Medicines Agency (EMA) | |
|---|---|---|
| Regulations | FDA 21 CFR Parts 210 and 211 include prescriptive requirements for documentation of validation activities. Part 210 deals with the manufacturing, processing, packing, or holding of drugs, while Part 211 deals with finished pharmaceuticals and is the foundation for process validation. | Volume 4, Annex 15 of “The rules governing medicinal products in the European Union,” is a 2015 guidance published by the European Commission covering principles of qualification and validation for the manufacture of medicinal products |
| Guidance | You’ll also want to download the 2011 guidance document on pharma process validation published by FDA. It covers:
|
What is the difference between pharmaceutical validation and qualification?
These often get confused. Qualification is a subset of validation. It is smaller in scope, more static in nature, and often includes tests performed on a single element or component of the pharmaceutical process.
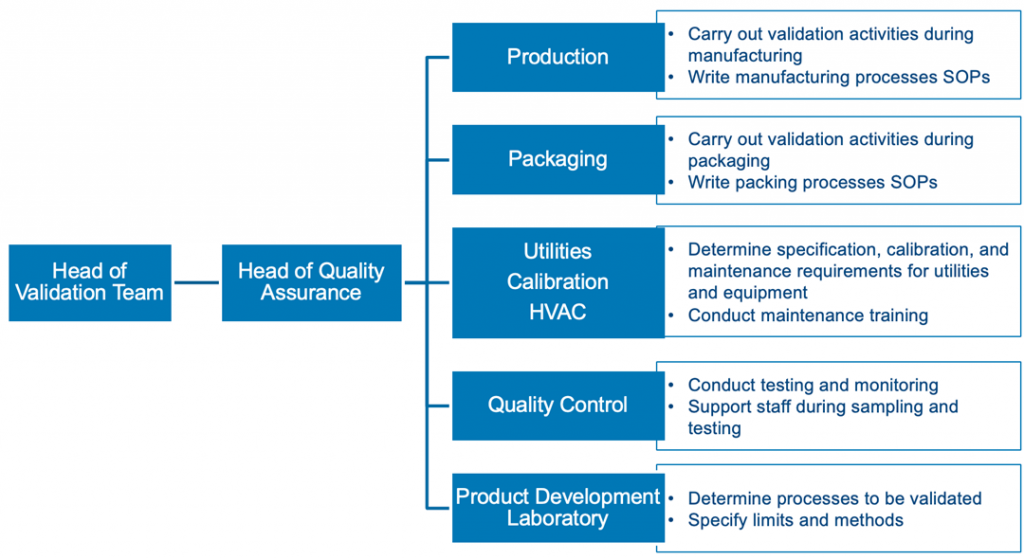
Pharma process validation isn’t a solo project. You need to get input from experts who know the drug and production process inside and out. Typically, your team will include members from quality assurance, engineering, industrial pharmacy, analytical chemistry, microbiology, and manufacturing. However, if your company is larger, it may also include people working in R&D, clinical engineering, purchasing, lab/technical services, or regulatory. Resist the urge to make the team larger than it should be. To keep things efficient, add people carefully, ensuring they have expertise in the process and can add significant value to the team. Below is an example of the team structure. Roles and responsibilities should be laid out in the validation master file.
Reading and doing are two entirely different things. If you are to be tasked with “doing” an actual validation project, consider our pharma validation training class. You’ll get an in-depth look at the entire pharmaceutical validation cycle, writing protocols, determining key operating parameters, establishing ranges for critical quality attributes, and identifying data analysis strategies. You will learn how to interpret the regulations, standards, and guidance documents; correctly apply the principles of risk management; create qualification protocols (DQ, IQ, OQ, PQ); and identify statistical methods and tools for implementing and maintaining process validation activities.

US OfficeWashington DC
EU OfficeCork, Ireland


