

Design validation focuses on the device itself and involves creating evidence that it meets user needs and intended uses. Process validation, as the name implies, focuses on the production of the device. Process validation demonstrates that, when a process is operated within specified limits, it will consistently produce product complying with established specifications and requirements. Most companies follow FDA requirements for design control 820.30 and ISO 13485 standard clause 7.3, and then perform validation during the final stage(s) of the product and process development sequence. However, planning begins with the initial design control process implemented by R&D. This helps to identify device quality characteristics and process variables.
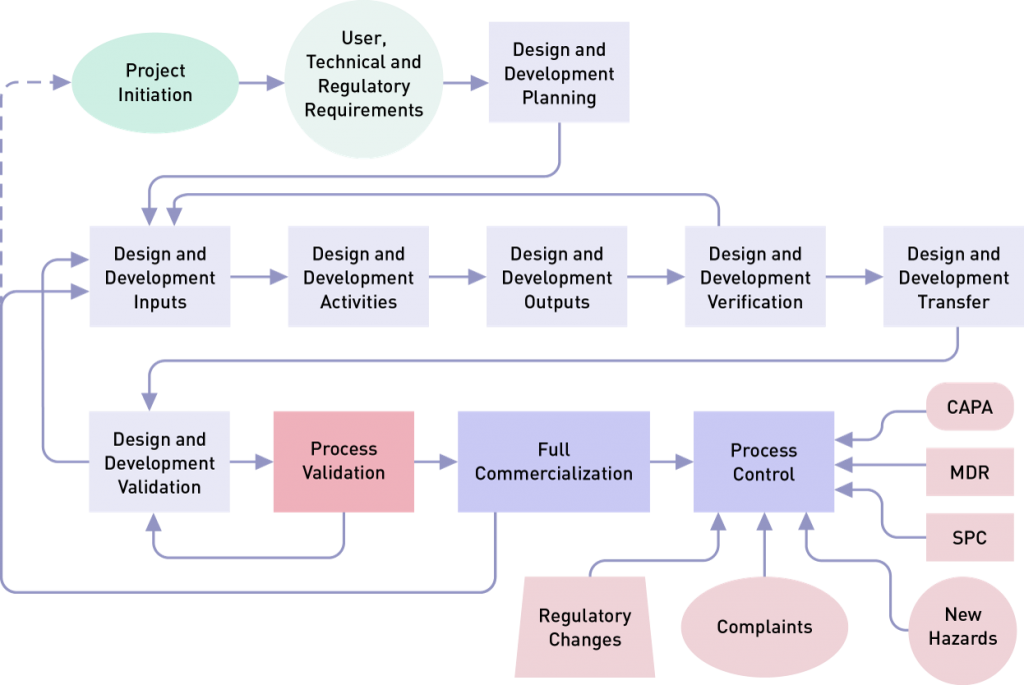
Note: Design and development planning is usually a project management type of activity, and design validation in many companies is a cevaluation activity.
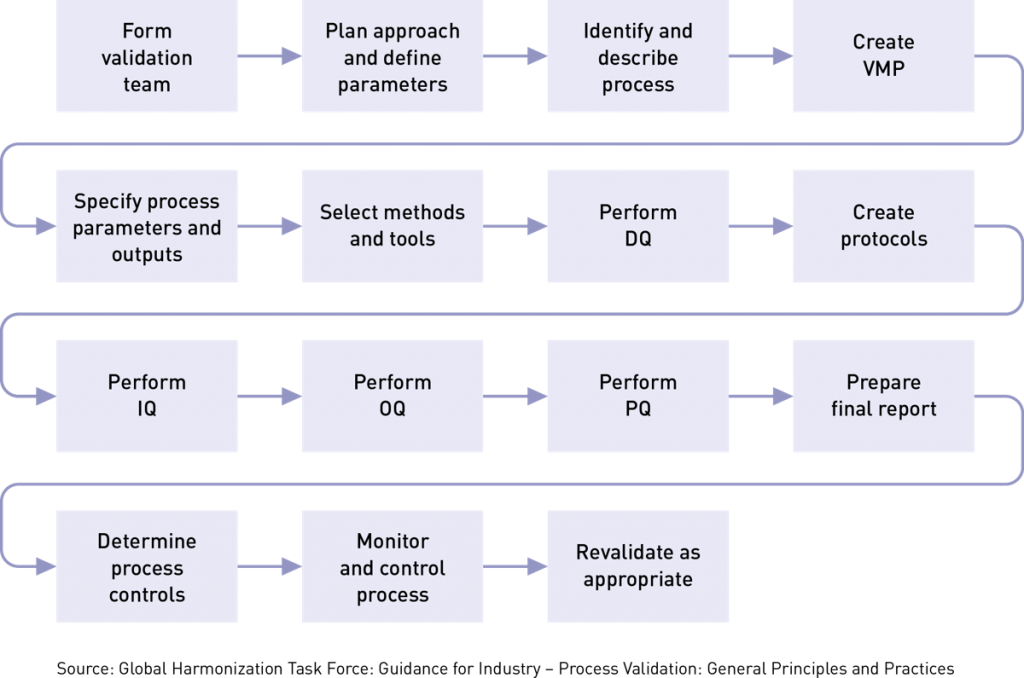
The basic principles of process validation must:
To ensure that a manufacturing process will consistently meet certain parameters, you must follow a systematic series of steps, such as those shown below. Some of these steps may be combined, but we have broken them out separately for clarity.

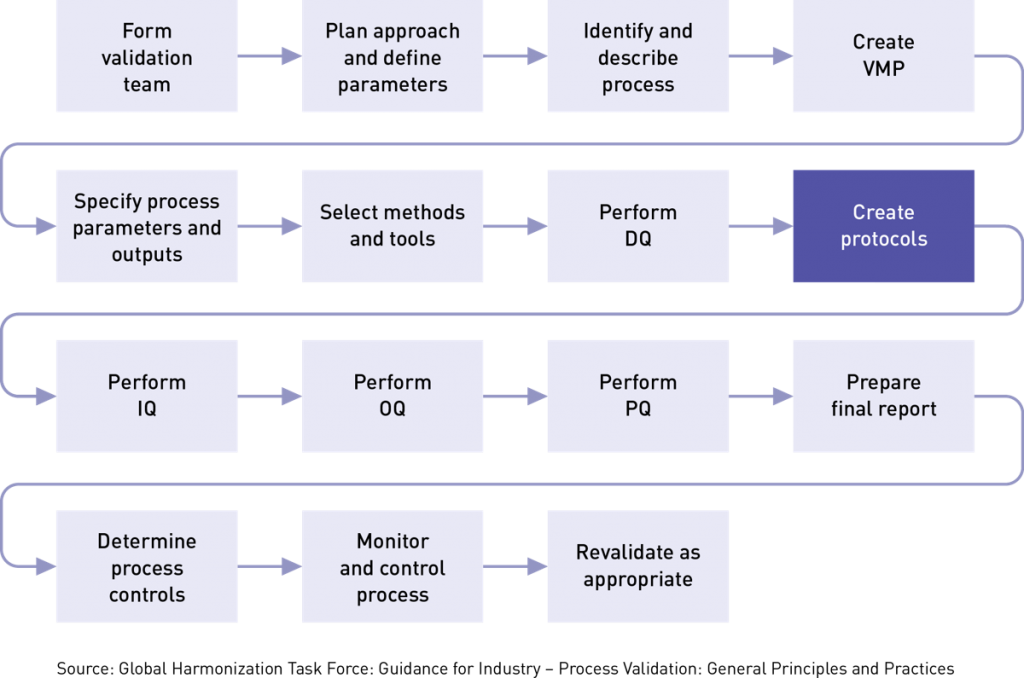
Your process validation plan provides a general framework for where you want to be, but your protocols are the actual maps on how to get there. Protocols are critical because they help determine if rules or procedures are done correctly and prevent crucial steps from being overlooked. They specify instructions or guidelines on how you plan to carry out a comprehensive study to investigate consistent operation of a new system or new equipment or procedure. Typically, protocols include significant background information. They explain the rationale for an objective of the study, give a full description of the procedures to be followed, set out parameters to be measured, describe how results will be analyzed, and provide predetermined acceptance criteria for reaching conclusions. Protocols determine:
You need to determine the best documentation strategy for your project. A complex piece of equipment like a filling line or a CMC will likely need a process validation plan that identifies the need for separate IQ, OQ, and PQ protocols. A simpler process/equipment such as a pH meter or balance may have a strategy that combines IQ, OQ, and PQ into a single plan/report.
It is important to reiterate that in order to write an effective protocol you need to fully understand the exact product requirements. That’s because your protocols will also establish your criteria for acceptance or rejection and outline the specific documentation you need. Remember, both the US FDA and ISO 13485 require you to document the results of your process validation activities, and this includes writing a clear, simple conclusion!
IQ can be executed by the facilities, engineering, production, or operations groups. The basic idea is to ensure that everything is installed properly. During this qualification phase, the installation of equipment, piping, services, and instrumentation will be checked against engineering drawings, piping and instrument diagrams (P&ID), and functional specifications developed during the project planning stage. During the project planning stage, all system elements, service conduits, and gauges are identified, and a documented record is prepared showing that all installed equipment satisfies your requirements. At various stages during IQ, you will need to review, check, report, and authorize protocols, documentation, procedures, equipment, specifications, and acceptance criteria for test results.
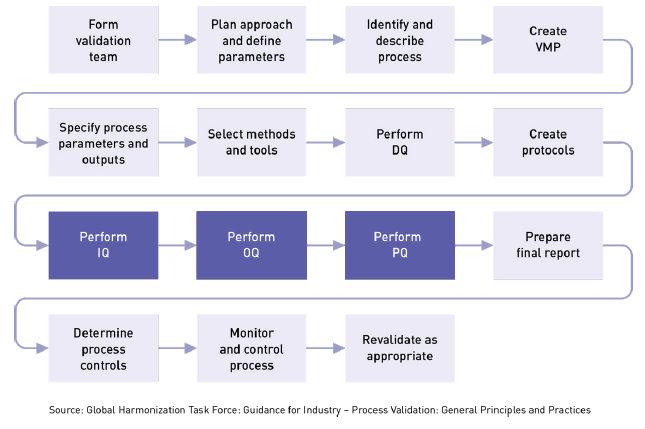
IQ touches on many tangible and intangible aspects related to installation, including:
There should be an SOP, checklist, or some other documented process that defines the standard installation procedure for each type of system or deliverable being installed. This is required for any equipment used in the manufacturing process. IQ verifies and documents that key aspects of an installation meet approved requirements. These requirements may come from:
When you buy a new car you don’t expect it to run smoothly forever. Likewise, despite your diligence and best efforts, glitches will occur and process refinements will be made. Even new or modified processes falter after implementation. Thus, it’s important to remember that process validation is never complete for long. Process performance must be monitored and maintained over time to ensure consistent performance, and full or partial revalidation of IQ, OQ, and PQ is simply part of the ongoing cycle. Revalidation requirements should always be considered an integral aspect of an original validation approval.


US OfficeWashington DC
EU OfficeCork, Ireland


