
Short answer: No.
Longer answer: From a high-level perspective, the standards now differ in intent, content, and structure.
The standards share one ultimate goal: Both ISO 9001:2015 and ISO 13485:2016 specify quality management system (QMS) requirements by which an organization must demonstrate that it can consistently provide products and services that meet customer and statutory/regulatory requirements.
ISO 9001, however, adds enhancing customer satisfaction and continual improvement as goals. As ISO 13485 has matured, it has moved way beyond ISO 9001. For the 2016 version, one objective of the working group that developed the new standard was to ensure ISO 13485 would better support the global alignment of regulatory requirements for medical devices.
ISO 9001 specifies requirements that are generic so that any organization, regardless of the products or services it provides, can use the standard. ISO 13485 specifies requirements for organizations involved in one or more stages of the medical device life cycle. To a large extent, ISO 13485 keeps the fundamentals of quality management systems we see in ISO 9001, adding or subtracting, where appropriate, requirements that are/are not relevant to medical devices.
There are also more definitions in ISO 13485, which makes sense when you consider that ISO 13485 serves a distinct, high-risk industry: medical devices.
While ISO 13485:2016 maintains the same structure of its prior version (based on ISO 9001:2008), the new structure of ISO 9001:2015 has an entirely fresh look and feel.
ISO 9001:2015 is based on Annex SL, which is intended to provide a uniform frame for writers to use when developing or revising standards. One goal of Annex SL is to support consistency in the writing of standards to make it easier to integrate multiple systems (e.g., quality, environmental, or safety).
The 2016 edition of 13485 does not follow the Annex SL structure, but it is possible that future editions of ISO 13485 may do so.
Below, a visual comparison between the two structures:
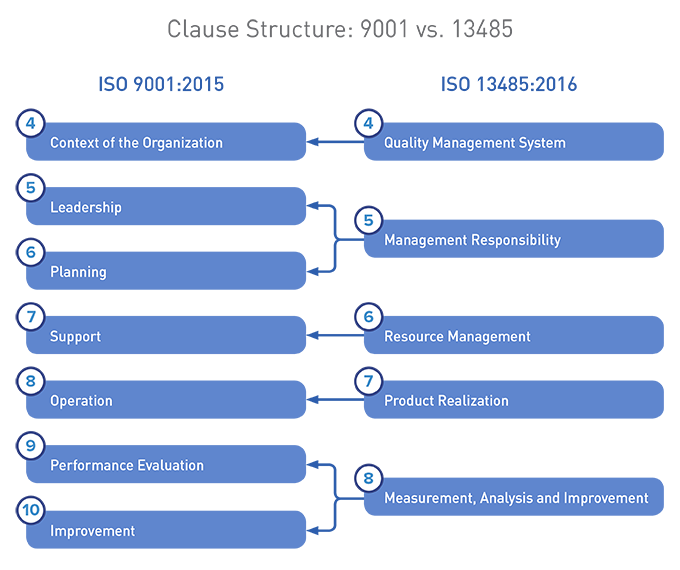
We maintain that ISO 9001:2015 and ISO 13485:2016 are not as aligned as you may have heard. Organizations holding certifications to both standards need to identify and address these differences as they upgrade to the most current versions of the standards.

US OfficeWashington DC
EU OfficeCork, Ireland


