QA/RA Consulting, Auditing & Training

Covered in this guide:
Real Purpose of the Medical Device QMS Audit
Basic Types of ISO 13485 Audits
Developing Your Overall ISO 13485 Audit Schedule
Preparing for Your ISO 13485:2016 QMS Audit
Conducting a QMS Documentation Review
Role of the ISO 13485 Lead Auditor
Creating the QMS Audit Plan
Ensuring Day 1 of Your Audit Goes Smoothly
Average ISO 13485:2016 Audit Duration
Conducting the Audit and Avoiding Rabbit Holes
Audit Interviewing Tips
Recording and Discussing Your Observations
Conducting the Closing Meeting
You Successfully Finished Your Audit. Now What?
How Much Detail Goes into the Final Audit Report?
ISO 13485 Internal or Supplier Audit Follow-Up Activities
Congratulations! You have been chosen (or perhaps conscripted) to conduct or participate in an ISO 13485 internal quality management system (QMS) audit. For many, the prospect of coordinating and conducting an audit can be overwhelming. However, believe us when we say the fear subsides with each hour of planning you do. In this white paper we will talk about how you can lay the foundation to ensure that your ISO 13485 audit progresses smoothly, yielding input that’s useful to your company’s management review as well as its corrective and preventive action (CAPA) processes.
Even though it seems obvious, it’s worth repeating that the purpose of conducting an audit is to determine whether the QMS conforms to specified requirements and is effective in enabling your organization to meet quality objectives. In other words, you are trying to assess whether the organization’s system says what it needs to say, that you’re doing what you say you’ll do, and that what you’re doing is working to produce the outcomes you need. A QMS audit is NOT intended to evaluate the quality of products, nor does it focus on the performance of people. The emphasis is on the QMS processes and the effectiveness of the entire system in meeting defined requirements and objectives.
Audits are planned, systematic processes carried out according to prepared working documents and audit plans.
ISO 13485 talks about two main components of internal audits (section 8.2.4):
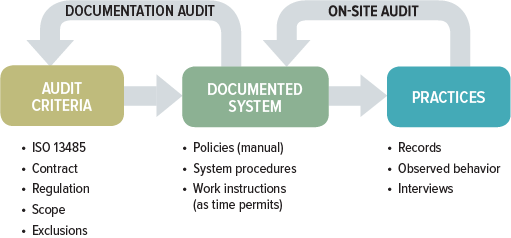
While documentation and on-site audits may seem like two entirely different animals, they are not. A thorough QMS audit includes both components. The difference between the two usually is in the approach and depth to which each of these audit components is conducted. The focus of the documentation audit centers on whether the QMS has been established and documented, while the on-site audit looks at whether there is sufficient objective evidence within the QMS to confirm it has been implemented and maintained.
A full QMS audit has four primary goals:
A well-planned audit schedule will ensure that audits are performed regularly, are conducted according to the importance of the process and address the results of previous audits.
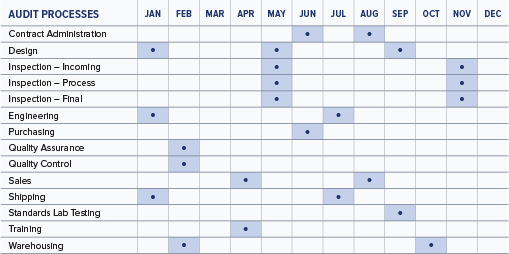
Developing a master audit schedule is the first step toward planning audit activities for the year. Individual audit leaders will construct the individual audit plans to meet the schedule. An example of a master internal audit schedule is shown below. A similar one could be developed to plan your supplier audits for the year.
A typical ISO 13485:2016 internal audit will generally cover 2-4 areas of the organization each month throughout the year, depending on the size of the company.
When planning an audit, it is tempting to skip some of the steps below and go immediately to creating a checklist and schedule. However, the process of initiating the audit is vital to assure the audit process is comprehensive and successful.Here are the steps you should take.
The purpose of the documentation review is to determine whether or not the QMS has been established and documented and meets the established regulatory requirements. Accordingly, where possible, try to review all documentation before the on-site audit activities commence. This will help you prepare for the on-site audit effectively and efficiently. Typically, auditees are required to submit a quality manual and procedures before the on-site audit.
The documentation should cover relevant information regarding the QMS (e.g., scope, exclusions that may exist) and any additional requirements beyond ISO 13485 and applicable regulatory requirements (e.g., customer requirements and/or supplier agreements). It should represent the documented quality management system as required by ISO 13485 in paragraphs 4.2.1 and 4.2.2 or other applicable criteria. If you are auditing a supplier, sometimes it might not be possible to get the quality manual ahead of time for proprietary reasons. If that’s the case, allocate time for a review at the beginning of the on-site audit. Organizational charts are helpful, so make sure you get a copy.
In addition to the manual and procedures, review:
Every audit has a lead auditor even if it’s the only auditor! This person represents the team in communication with the auditee and management. The lead auditor also defines the requirements of each audit assignment, including qualification of other audit team members. Here are some of the lead auditor’s additional responsibilities:
Starting an on-site audit without a detailed plan is a surefire way to waste a lot of time, frustrate a lot of people, and leave without generating useful output. In an ideal world, you should spend 2 hours planning every hour of audit time. A detailed audit plan should cover:
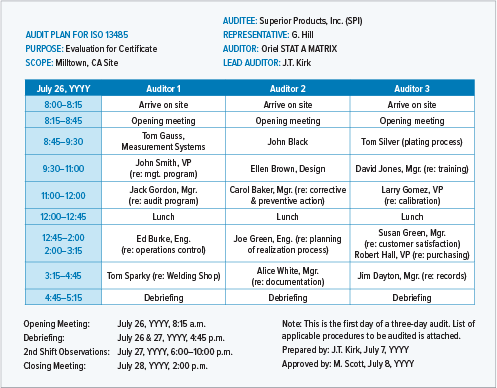
This is an example of an internal audit plan for a single internal process.

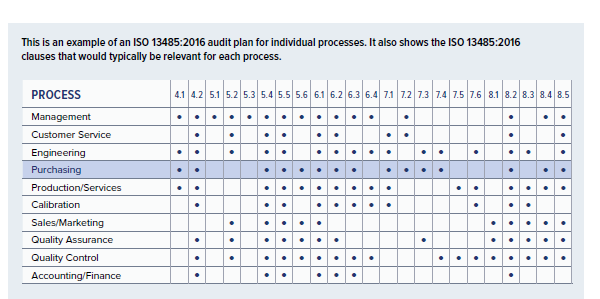
This is an example of an ISO 13485:2016 audit plan for individual processes. It also shows the ISO 13485:2016 clauses that would typically be relevant for each process.
An essential part of the audit planning stage involves preparation of the working documents. You’ll usually do some of this in parallel with the documentation review portion of the audit, which will give you information about specific topics and information paths to follow during your on-site audit.
Working documents typically include checklists, audit sampling plans and forms for recording meeting attendance, audit evidence, and audit findings (corrective action reports, nonconformity reports). Checklists are good tools, as they save valuable time and ensure that important items are not missed during the audit. It is worth spending time on these, because checklists can be adapted for use in other audits and improved based on your experience over time. Just remember: As you’re auditing, don’t use checklists like a script; instead, consider them only as a guide. Also, don’t forget to safeguard and treat your audit documents as confidential or proprietary at all times.
The final step in the preparation phase is to confirm the audit details with your auditee. This correspondence comes from the lead auditor and must follow company procedures and address all points from any previous phone discussions, meetings, or emails. The notification must confirm the date, time, and place of the opening meeting and include the audit plan and proposed schedule/agenda. (Optionally, you could include a copy of your checklists if they will aid understanding, but there are pros and cons to doing so.) The purpose of this notification is to ensure there are no misunderstandings.
A detailed audit plan will be very specific about times, participants, and process areas.
You have spent weeks preparing for your audit. All documentation has been reviewed, schedules created, auditees notified, and checklists confirmed. Now it’s time for the scary part: Conducting the audit! If you have done your job well to this point, the audit should be the easy part because you will simply be executing a well-choreographed plan.
On the morning of Day 1, you will host the opening meeting. There are many things you will want to accomplish during this meeting, including:
The duration of an audit is based on the number of employees in the facility and the scope of the QMS. The risk associated with the device is also a factor. For example, there is certainly more risk associated with manufacturing heart valves than manual wheelchairs, and this impacts audit length. The International Accreditation Forum documents MD-5 and MD-9 set guidelines for internal audit days as well as general protocols for conducting an ISO audit. It should be noted, however, that this type of audit length determination is trending out with the use of audit duration calculations used in the Medical Device Single Audit Program Model (MDSAP). MDSAP audits are based on the number of elements to be covered in the audit. These types of audits can be considerably longer than an ISO audit.
All that preparation you did in the weeks leading up to the audit will now pay off. You should make every effort to deal directly with the people involved in implementing the system. People – not documents – make or break a system. When you start performing the audit, it is important to remember that an audit is really a method of sampling and is conducted to get a sense of what is happening. Consider stratified random sampling to focus the audit based on risk (e.g., rather than taking a random sampling of purchase orders, stratify the population by criticality to focus on what is important). You need to be sure that the auditee is not cherry-picking documents to show you. You should dictate the documents you want to see, reviewing the requisite number of samples stipulated in your audit plan.
During the audit, you will invariably come across people who nervously ramble, digress, or are intentionally vague or evasive. In these cases, it is important that you remain courteous but persistent. Be polite but insist on getting details needed to answer the question. Don’t go down the rabbit hole with someone who is trying to explain something that is irrelevant. It is the auditor’s job to keep the auditee on track and extract the information needed. That being said, you should explore problems fully. Accordingly, you may need to go beyond your checklist to dig deeper and look at key process interactions that may be relevant (e.g., purchasing and production interaction).
Auditees often get nervous during an ISO 13485:2016 audit because they sometimes feel as though they are being personally interrogated. To gain their cooperation, it is important that you set a commonality of perceived purpose in the opening meeting. Your common goal is to ensure that the company has a quality management system that is conforming to requirements and effective, not to throw someone under the bus. Make sure to tell the auditee that you will be taking notes during an interview. Refer to your checklists repeatedly but don’t read verbatim from them; instead, use the checklist items as a framework for discussion. To get relevant, complete information from auditees,
follow these guidelines:
Remember, although the audit may be the most important thing in your professional life at this moment and you may feel like the most powerful person in the room, your presence is an imposition for the auditee. They have other work to do. With limited time to collect the information you need, think carefully about how you ask questions. Consider these alternative examples:
The second question (i.e., an open question) is likely to reveal much more information about who, what, when, where, why, and how revisions are issued. Also, keep personnel dynamics in mind. Auditee personnel may hold back information if their boss is also in the room.
Audits can be exhausting, and you’ll be eager to go home at the end of a long day. Resist the urge! It is vital that you conduct a debriefing at the end of each day (not the next morning) to discuss observations with your audit team members and ensure that team members are performing their assigned functions. Document your observations so each team member can evaluate results for potential nonconformities. Also, you’ll sleep better that night with all of your insights safely put on paper instead of cluttering your brain.
Don’t meet only with your audit team. It is important that you keep the auditee fully aware of what is being observed. Meet with the auditee per an established schedule for debriefing and report good as well as nonconforming conditions.
When the audit is complete, the audit team will conduct a closing meeting with the management team to formally present positive findings, cite concerns, share opportunities for improvement, and clarify misunderstandings. This meeting and the final ISO 13485 audit report are critical to the success of the audit, so the lead auditor must be fully prepared with notes covering all areas and have supporting objective evidence for each finding.
The purpose of the closing meeting is to present logical and fact-based explanations of the strengths and weaknesses of the quality management system. You will want to explain to management that the audit investigated only a sample of activities and that there may be other nonconformities the sampling did not uncover. This is especially important for people to understand because an actual FDA inspection or Notified Body audit may uncover different issues. You don’t want people pointing fingers at you if observations arise that were not revealed by an internal audit.
With regard to nonconformities, it is best not to raise these for the first time during closing meetings. Always bring the issue up during the audit and give the auditee an opportunity to explain something you may have misunderstood. If there is still evidence of a nonconformity, let the auditee know then. Also, make sure you give credit where credit is due, particularly in areas where procedures have been shown to be effective. When covering deficiencies, focus the auditee’s attention on the significance of the nonconformities (major versus minor). Get agreement on a timeframe for creating a corrective action plan, and a deadline for addressing those deficiencies. You should also state the date when the final audit report will be issued. Finally, although not required (especially with internal audits), it’s a good idea to keep minutes of the meeting and record attendance.
You’ve spent weeks preparing for your audit and several days conducting it. Now comes the time to formally put your thoughts and findings on paper. The purpose of the audit report is to present the auditee with a written record of nonconformities and provide a full account of audit evidence that supports these nonconformities. In general, your audit report should:
Don’t forget – your report should not contain surprise nonconformities that were not discussed during the audit and in the closing meeting.
The nature of the audit will determine the characteristics such as the length, format, emphasis areas, and sequence. Nonetheless, the formal report should contain a highly detailed description of the quality management system’s strengths, nonconformities, audit evidence, opportunities for improvement, and areas of concern. It should include:

The content of the ISO 13485 QMS audit report must represent the conclusions of the lead auditor with input from the entire audit team, and not just the viewpoints of individuals. This gives the auditee the benefit of the collective experience of all team members and reduces bias.
The lead auditor will decide if the scope of the audit warrants including corrective action requests in the final report. Your audit report should be sent to the auditee as soon after the closing meeting as practical. This is important because it reinforces the points you made during the closing meeting and keeps those issues top of mind with the auditee management team.
Now that you’ve crafted a beautifully detailed report and submitted it to the auditee, you’re finished – right? Not so fast. The last thing you want is to show up at the next audit only to find out that nothing has been done to address nonconformities described in your audit report. Inaction would certainly frustrate you and it would not be good for the company. Thus, after the closing meeting has occurred and the audit report has been sent to management, your goals are to:
It’s also a good idea to make sure the organization has a methodology to address corrective actions. If not, this would be a good opportunity for improvement. Without a methodology supported by tools, chances are that the CAPA system will not be effective.
As part of the follow-up process, you should also retain or destroy documents pertaining to the audit in accordance with any agreements, procedures, and applicable statutory, regulatory, and contractual requirements.
As an auditor, you play a critical role in the health of your organization’s quality management system, and ultimately the safety of the medical devices your company produces. That’s an important responsibility, which needs to be taken seriously. The benefits of sustained audits are much the same as eating healthfully or exercising. It may not always feel great right away, but the long-term results are always positive.
This blog only scratches the surface of the topic. If you will be more involved in doing audits for your organization, we highly recommend you check out our ISO 13485 lead auditor training class, which offers the opportunity to become certified by Exemplar Global. Our team is also available to conduct internal and supplier audits as needed.

US OfficeWashington DC
EU OfficeCork, Ireland


