QA/RA Consulting, Auditing & Training

Unlike Regulatory Authorities in most other countries, the US Food and Drug Administration (FDA) relies on random inspections to keep medical device manufacturers honest.
Random audits can occur at any time, and that means manufacturers must always remain in compliance with the FDA Quality System Regulation (QSR).
There are simply too many medical device manufacturers for FDA to inspect annually, so FDA prioritizes its limited inspection staff according to the risk posed by manufacturers ,their devices and if the company has had any product related problems in the recent past. Some companies get more attention than others. In general, Class II and III manufacturers can expect an FDA visit every two years. The risk-based approach means companies with new or existing Class III devices are the highest priority, especially those making implantable and life-supporting devices. Class II manufacturers (the bulk of the volume) are next in line, along with any companies that have recently introduced a device via the 510(k) process. Occasionally, FDA will inspect higher-risk Class I manufacturers. Companies that have had their hand slapped in the past tend to get frequent repeat visits, which are often unannounced. FDA can also conduct “for cause” inspections – hopefully you’re not reading this article to find out what that means.
If you have not yet had the pleasure of experiencing an FDA inspection, here’s how it goes down.
Generally, FDA will notify US-based manufacturers at least 5 days before an inspection. Foreign companies typically get more advance notice so that travel and translator arrangements can be made. Before the inspection, FDA may request that you send them a copy of your quality system manual and management review procedures to facilitate the inspection. You are not required to send these documents, but refusing to do so might put the inspector in the frame of mind to think that you have something to hide.
When the FDA inspectors arrive (there may be more than one, depending on the size of your company), they will present their badge and ask to see the most senior management official. While you are allowed to write down their name and badge number, you may not photograph or copy their badge. They will then present a Form 482 “Notice of Inspection” as a formality and have an opening meeting to learn more about the structure of your company and products, followed by a facility tour.
The FDA inspection approach focuses on reviewing procedures and then examining records associated with them. To understand what they will cover and how, study the 108-page QSIT manual prepared to guide FDA field staff and share it with others who might be involved in the inspection. It dates from 1999 but is still in use today.
The inspection process focuses on four main subsystems (as shown in blue below) with the ultimate question being: Did the management team provide the necessary resources to establish and maintain an effective QMS? The other three subsystems are reviewed via links with the four main subsystems. All seven subsystems orbit around management controls
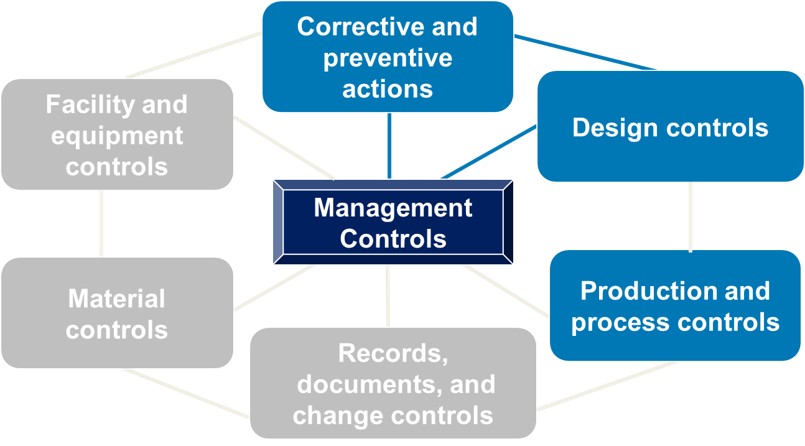
If this is your first FDA audit, the inspectors will cover all four subsystems. During subsequent normal inspections, the inspectors will cover CAPA plus one additional subsystem. As explained in a 2019 draft guidance document from FDA, the typical inspection can be expected to last 3-6 business days. FDA should tell you how many days they expect the inspection will last when it notifies you of the impending audit. Remember, the length of time may depend on what the FDA finds when onsite.

The process is top down. It begins with a review of your procedures and then dives into a random sampling of associated records. (If you love stats and want to get into the nuts and bolts of how many samples FDA will review, you’ll enjoy pages 103-107 of the manual.) The FDA inspectors don’t have the authority to demand to see your internal audit reports, supplier audit reports, or your management review meeting minutes. They can request them, but you it is up to the organization whether they are made available. This should be defined in an established procedure within the QMS. However, they will check to make sure you have these documents. While the inspection is quite methodical, as outlined in the table below, the inspectors may dig deeper if they find an area of concern. As they proceed through the inspection, they will point out Form 483 observations in real time. These are more serious areas of concerns than “discussion items,” which get documented in the final report but could later be elevated to observations. You can expect that the FDA inspectors will hold daily meetings with the management team to disclose what was discovered that day.




At the conclusion of what is sure to be an exhausting and stressful week for you, the investigators will meet with management to review all observations and discussion points. The inspectors will give management an opportunity to defend or clarify their position and will document everything in an Establishment Inspection Report (EIR). The date and status of your inspection (but not the actual EIR) will be posted on the FDA website within a week. The most important field here is “Classification,” which is separated into three categories:
NAI – No Action Indicated (inspection went well, this is what you hope to see)
VAI – Voluntary Action Indicated (the inspectors found some problems; FDA expects the company to take corrective action)
OAI – Official Action Indicated (the inspectors found serious issues; this means a 483 will likely be issued)
Of course, the outcome shouldn’t be a surprise to you, as the FDA inspectors would have pointed out problems throughout the inspection and in daily meetings. You can request a copy of your EIR within 60 days. If things did not go so well, the inspectors may issue an FDA Form 483 and you will have 15 days to respond. FDA could take things to the next level by issuing a Warning Letter, but this is increasingly rare. By the way, when the FDA investigator presents the 483, you are NOT required to sign it. If you do, that’s an admission that everything in the report is accurate and true in a court of law….if it comes to that. Again, whether you do or don’t sign should be documented in a formal procedure in your QMS.
If you made it this far, there’s a good chance that you are heavily involved in FDA QSR and ISO 13485 compliance. If so, consider our in-depth training on ISO 13485 and FDA QSR, or our ISO 13485 lead auditor training class. Alternatively, ask us about having one of our skilled auditors conduct outsourced audits on your behalf.

US OfficeWashington DC
EU OfficeCork, Ireland


