
FDA uses a risk-based predicate-based review approach. This means that when you submit your application to FDA, you will be comparing your medical device to a very similar device that has already been approved (the predicate) by FDA. Because FDA requires you to identify a single predicate device, your first step will be to find one. You may already have a good idea of which competitive products would make a suitable predicate for comparison in your 510(k). In any case, you should start your research using the FDA Product Classification database.
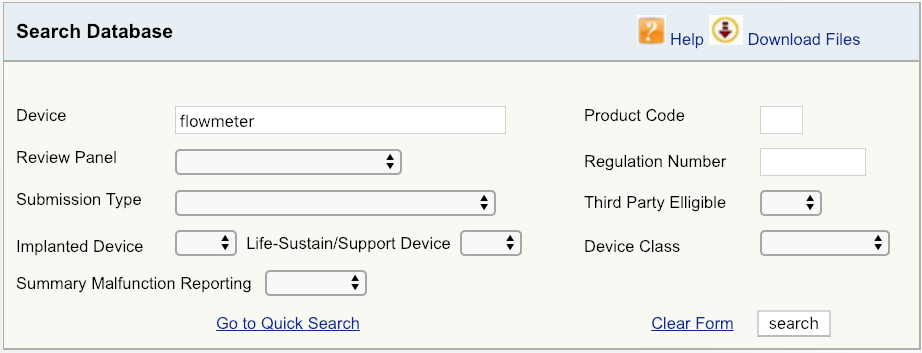
For example, let’s say your company is introducing a new cardiovascular blood flowmeter to the US market. The first step would be to begin with a simple device search on the FDA database, as shown, and then look at the options available. Start with broadest definition of your product – in this case, just the term “flowmeter.” The results show that there are six unique FDA product codes for products related to flowmeter.
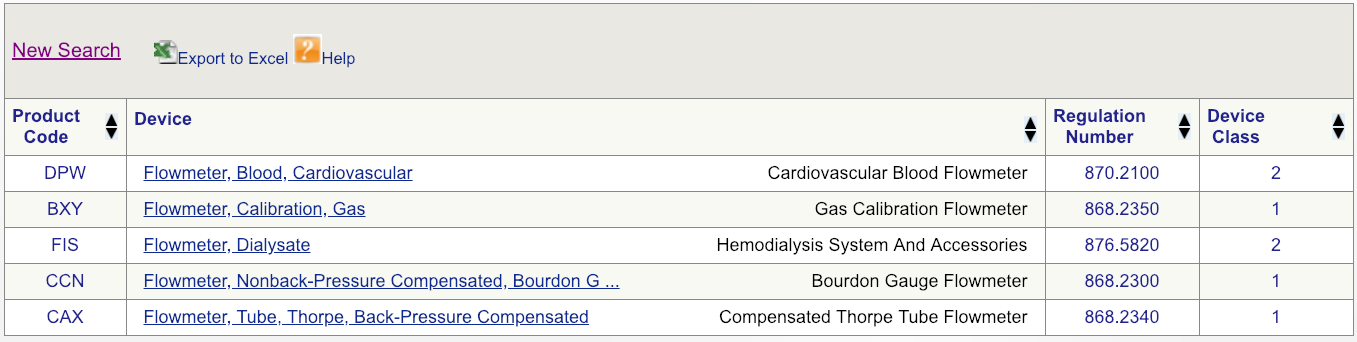
Code DPW looks to be the best match but, to make sure, click on the regulation number and carefully read the description.
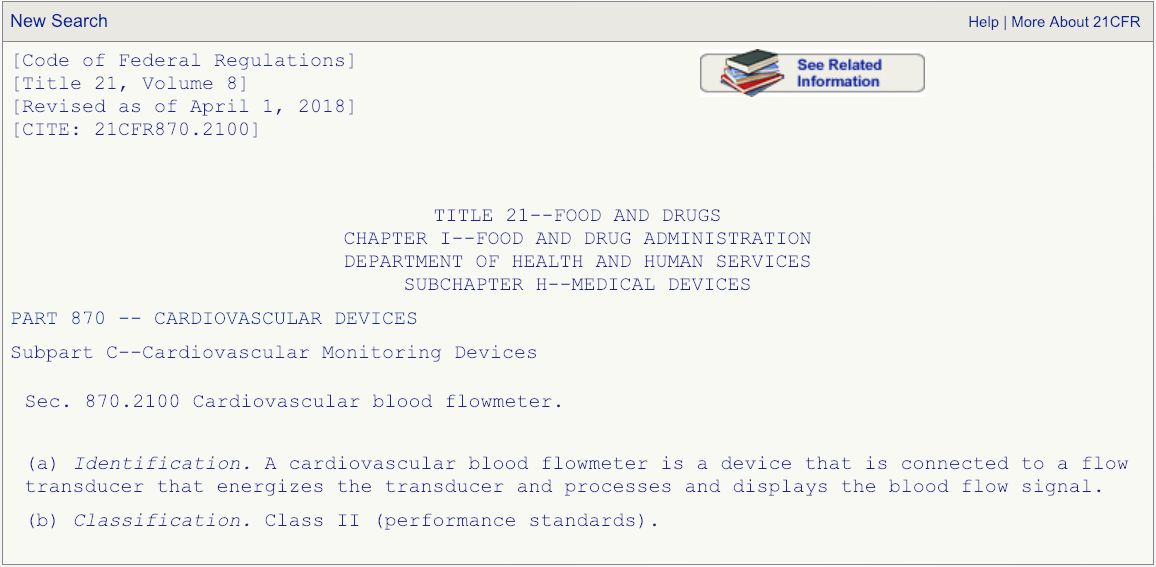
After you have read the description associated with the regulation number and are absolutely certain that the product code DPW is the correct one that fits your device, then go the FDA’s 510(k) database and search for any devices cleared under product code DPW.
This is where things can get tricky and you need to be careful. In this example, there are 131 cleared medical devices under classification product code DPW. Which one will make the best predicate for your device? Well, here’s a piece of advice: When reviewing your options (hopefully you will not have 131 options), it is best to sort by the “Decision Date” column and start with devices that were cleared recently. Why? While it may be tempting to choose an older device as your comparative predicate, the FDA frowns upon using devices cleared more than 10 years ago.
Your next step will be to click on the “Summary” link for each device as shown (see the example page below).
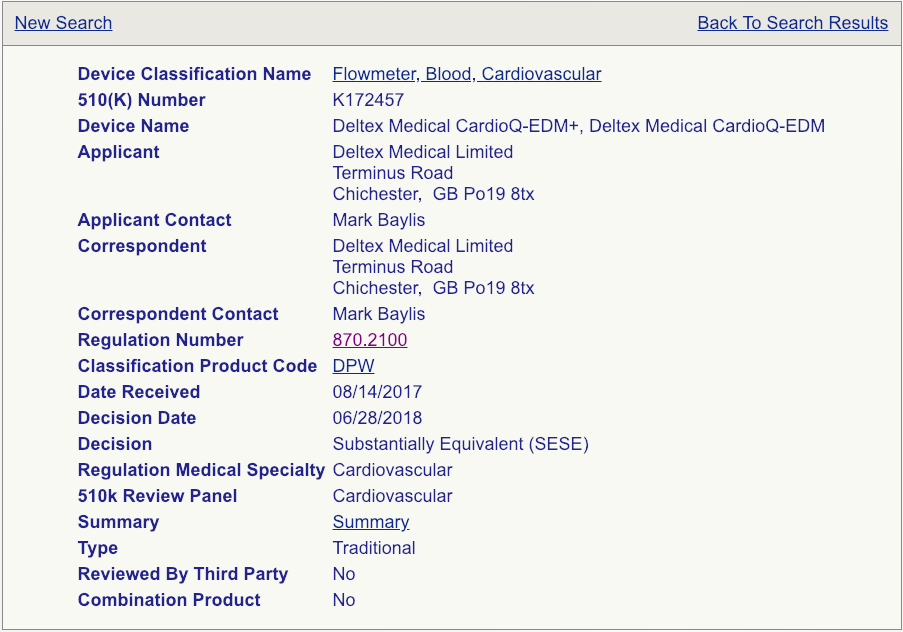
Read these summaries very, very carefully. Pay attention to the intended use, allowed indications for use, testing conducted, and clinical studies that may have been performed. Some 510(k) summaries provide more information than others, so make sure you review as many as possible and aggregate your knowledge in a spreadsheet if you are reviewing a lot of summaries. Your chosen predicate does not need to be identical to your device, but it needs to be close enough not to raise additional safety and effectiveness questions. The chosen predicate must have the same intended use and indications for use. This is very important. If the indications for use are different, that device won’t be a suitable predicate. The technological features should closely match your device. Choosing the right predicate is truly critical for the success of your submission and, if you have any reservations about your options, you should seek the advice of an experienced FDA consultant.
Plan on six months from the hopeful day you submit until the joyous occasion when you are holding that “substantial equivalence” letter in your hand. In all fairness, because such a high percentage of companies receive additional information requests from FDA, the amount of total time that FDA spends reviewing your submission is only slightly longer than the amount of time companies spend replying to FDA requests. The average time to clearance is around five or six months but that also varies by device.


US OfficeWashington DC
EU OfficeCork, Ireland


