Watch time: 2 min
In this video
What you need to know
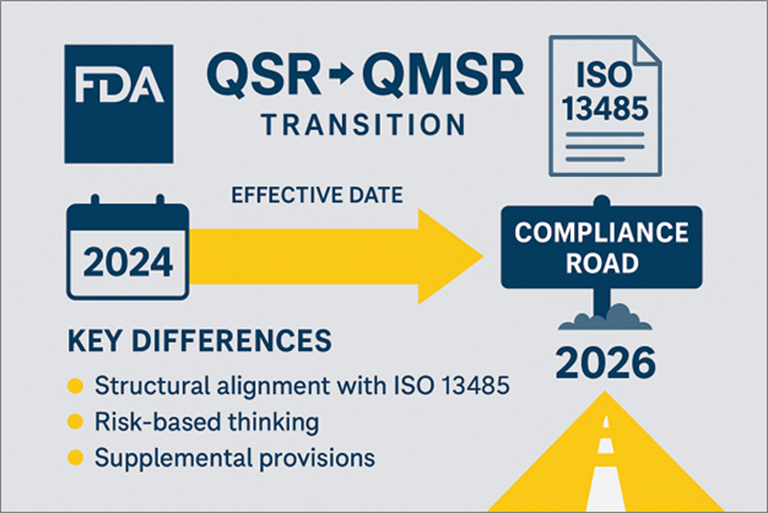
Risk management is no longer just a design-phase requirement – under QMSR and ISO 13485, it must be integrated across the entire product life cycle. Learn what this shift means and how to stay compliant by 2026.
As FDA transitions from QSR to QMSR, risk management is no longer just a box to check during design; it must be embedded throughout the entire product life cycle.
This video breaks down what this shift means, how ISO 13485 and ISO 14971 factor in, and what steps manufacturers need to take to stay compliant by 2026.
Watch time: 2 min
In this video
How do process thinking and risk controls drive better quality systems?
In our latest video, we break down two core pillars of QMSR (Quality Management System Regulation) for medical devices: the process approach and risk-based controls.
You’ll learn how the PDCA cycle helps teams plan, execute, and improve processes, and how risk-based thinking ensures consistency, accuracy, and regulatory alignment.
Whether you’re in quality, regulatory, or operations, this is a quick, clear look at what makes a QMS truly effective.
Watch now to see how these concepts come together for stronger compliance and smarter systems.